

Paroxysmal Movement Disorders
- PMID: 34177764
- PMCID: PMC8232056
- DOI: 10.3389/fneur.2021.659064
Paroxysmal Movement Disorders
Abstract
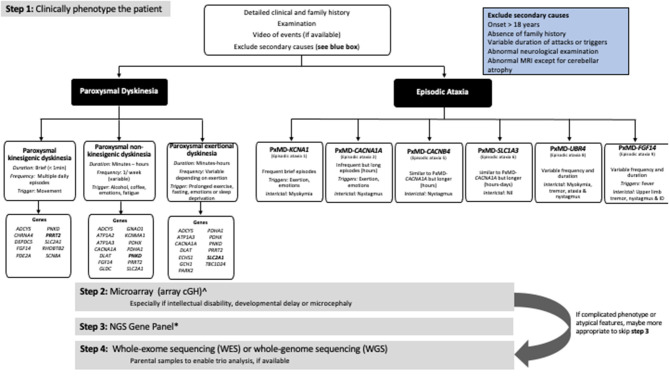
Paroxysmal movement disorders (PxMDs) are a clinical and genetically heterogeneous group of movement disorders characterized by episodic involuntary movements (dystonia, dyskinesia, chorea and/or ataxia). Historically, PxMDs were classified clinically (triggers and characteristics of the movements) and this directed single-gene testing. With the advent of next-generation sequencing (NGS), how we classify and investigate PxMDs has been transformed. Next-generation sequencing has enabled new gene discovery (RHOBTB2, TBC1D24), expansion of phenotypes in known PxMDs genes and a better understanding of disease mechanisms. However, PxMDs exhibit phenotypic pleiotropy and genetic heterogeneity, making it challenging to predict genotype based on the clinical phenotype. For example, paroxysmal kinesigenic dyskinesia is most commonly associated with variants in PRRT2 but also variants identified in PNKD, SCN8A, and SCL2A1. There are no radiological or biochemical biomarkers to differentiate genetic causes. Even with NGS, diagnosis rates are variable, ranging from 11 to 51% depending on the cohort studied and technology employed. Thus, a large proportion of patients remain undiagnosed compared to other neurological disorders such as epilepsy, highlighting the need for further genomic research in PxMDs. Whole-genome sequencing, deep-sequencing, copy number variant analysis, detection of deep-intronic variants, mosaicism and repeat expansions, will improve diagnostic rates. Identifying the underlying genetic cause has a significant impact on patient care, modification of treatment, long-term prognostication and genetic counseling. This paper provides an update on the genetics of PxMDs, description of PxMDs classified according to causative gene rather than clinical phenotype, highlighting key clinical features and providing an algorithm for genetic testing of PxMDs.
Keywords: episodic ataxia; genetics; next-generation sequencing; paroxysmal dyskinesia; paroxysmal movement disorders.
Copyright © 2021 Harvey, King and Gorman.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Genetic Links to Episodic Movement Disorders: Current Insights.Appl Clin Genet. 2023 Mar 1;16:11-30. doi: 10.2147/TACG.S363485. eCollection 2023. Appl Clin Genet. 2023. PMID: 36883047 Free PMC article. Review.
-
Paroxysmal movement disorders: An update.Rev Neurol (Paris). 2016 Aug-Sep;172(8-9):433-445. doi: 10.1016/j.neurol.2016.07.005. Epub 2016 Aug 25. Rev Neurol (Paris). 2016. PMID: 27567459 Review.
-
Paroxysmal Movement Disorders: Recent Advances.Curr Neurol Neurosci Rep. 2019 Jun 11;19(7):48. doi: 10.1007/s11910-019-0958-3. Curr Neurol Neurosci Rep. 2019. PMID: 31187296 Review.
-
Paroxysmal movement disorders: Paroxysmal dyskinesia and episodic ataxia.Handb Clin Neurol. 2023;196:347-365. doi: 10.1016/B978-0-323-98817-9.00033-8. Handb Clin Neurol. 2023. PMID: 37620078 Review.
-
16p11.2 deletion in patients with paroxysmal kinesigenic dyskinesia but without intellectual disability.Brain Behav. 2018 Nov;8(11):e01134. doi: 10.1002/brb3.1134. Epub 2018 Oct 11. Brain Behav. 2018. PMID: 30307717 Free PMC article.
Cited by
-
A Rare Cause of Paroxysmal Movement Disorder Associated with TBC1D24 Gene Mutation in Two Siblings.Ann Indian Acad Neurol. 2023 May-Jun;26(3):290-293. doi: 10.4103/aian.aian_465_22. Epub 2023 Apr 24. Ann Indian Acad Neurol. 2023. PMID: 37538433 Free PMC article. No abstract available.
-
Scoping Review on ADCY5-Related Movement Disorders.Mov Disord Clin Pract. 2023 Jun 6;10(7):1048-1059. doi: 10.1002/mdc3.13796. eCollection 2023 Jul. Mov Disord Clin Pract. 2023. PMID: 37476318 Free PMC article.
-
Exploring the Genetic Landscape of Chorea in Infancy and Early Childhood: Implications for Diagnosis and Treatment.Curr Issues Mol Biol. 2024 Jun 6;46(6):5632-5654. doi: 10.3390/cimb46060337. Curr Issues Mol Biol. 2024. PMID: 38921008 Free PMC article. Review.
-
Clinical characterization of a novel episodic ataxia in young working Cocker Spaniels.J Vet Intern Med. 2025 Jan-Feb;39(1):e17268. doi: 10.1111/jvim.17268. J Vet Intern Med. 2025. PMID: 39715410 Free PMC article.
-
Rare Movement Disorders-An Approach for Clinicians.Int J Mol Sci. 2025 Jun 23;26(13):6024. doi: 10.3390/ijms26136024. Int J Mol Sci. 2025. PMID: 40649801 Free PMC article. Review.
References
-
- Straub J, Konrad EDH, Grüner J, Toutain A, Bok LA, Cho MT, et al. . Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in drosophila. Am J Hum Genet. (2018) 102:44–57. 10.1016/j.ajhg.2017.11.008 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources