

Factor D Inhibition Blocks Complement Activation Induced by Mutant Factor B Associated With Atypical Hemolytic Uremic Syndrome and Membranoproliferative Glomerulonephritis
- PMID: 34177949
- PMCID: PMC8222914
- DOI: 10.3389/fimmu.2021.690821
Factor D Inhibition Blocks Complement Activation Induced by Mutant Factor B Associated With Atypical Hemolytic Uremic Syndrome and Membranoproliferative Glomerulonephritis
Abstract
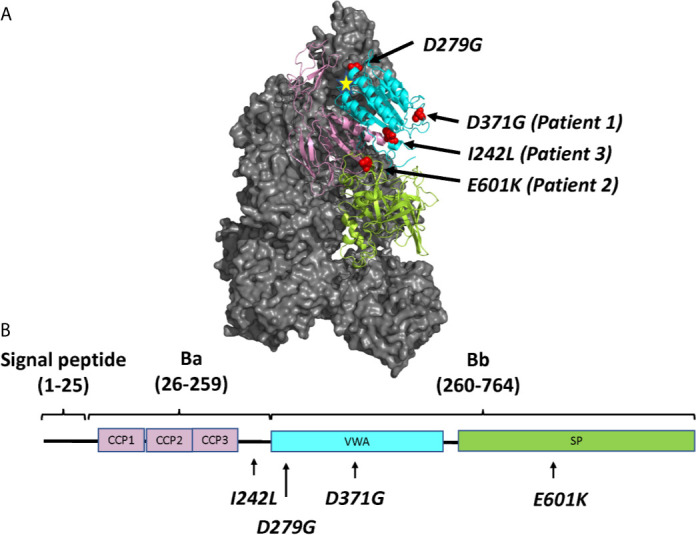
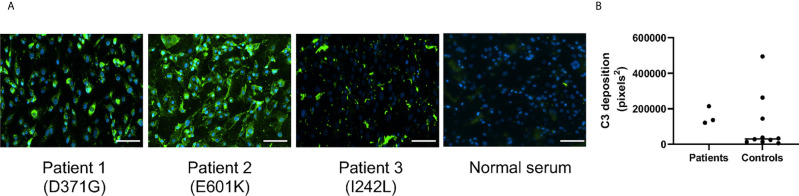
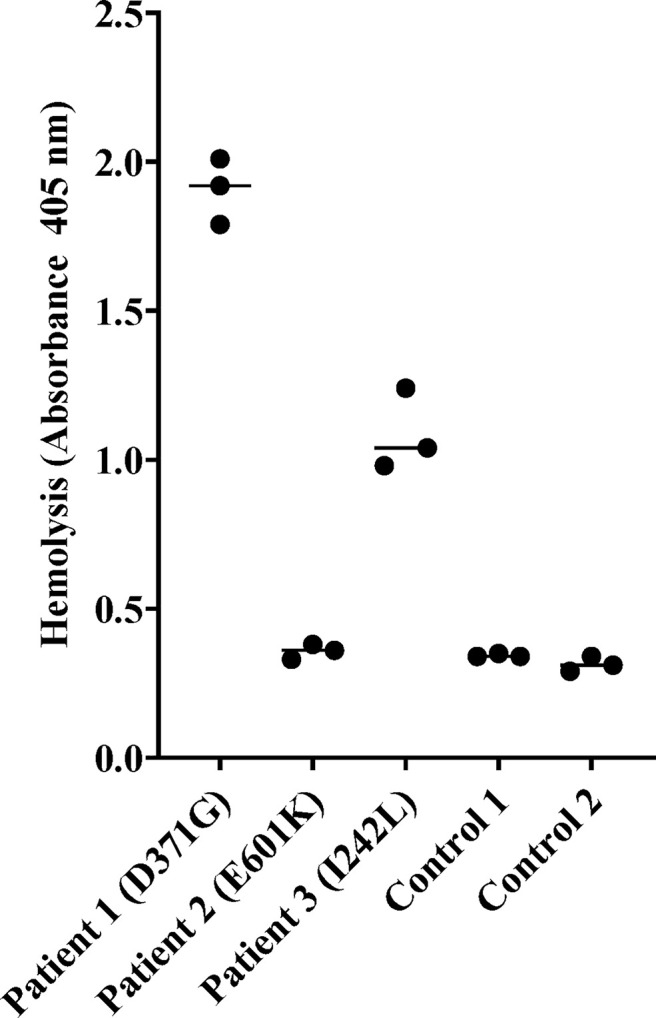
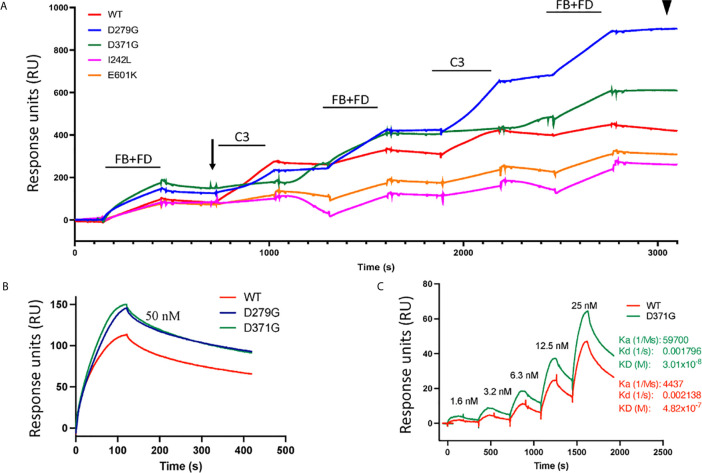
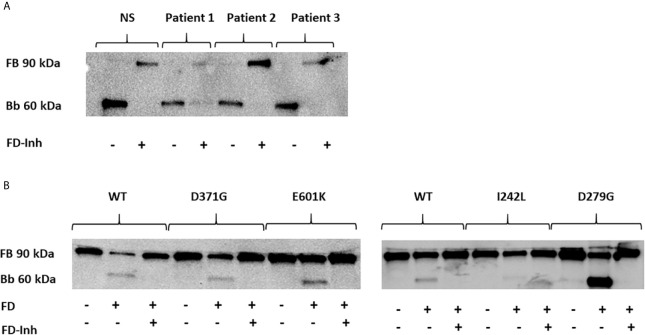
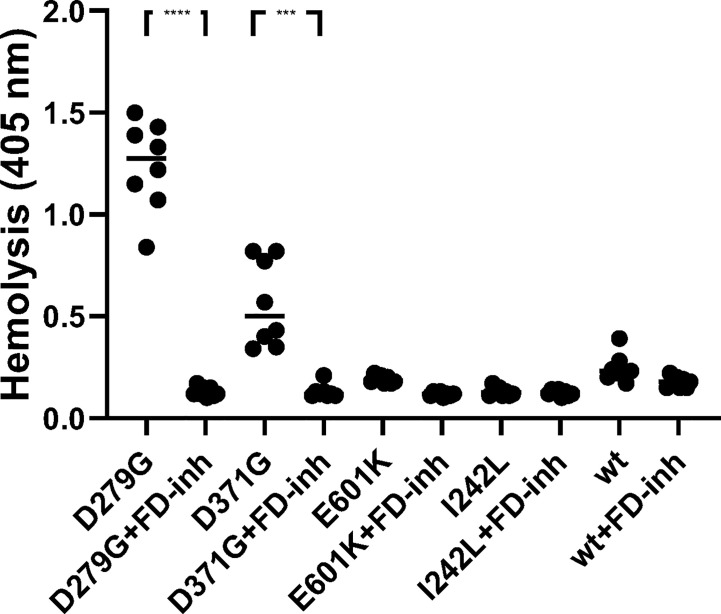
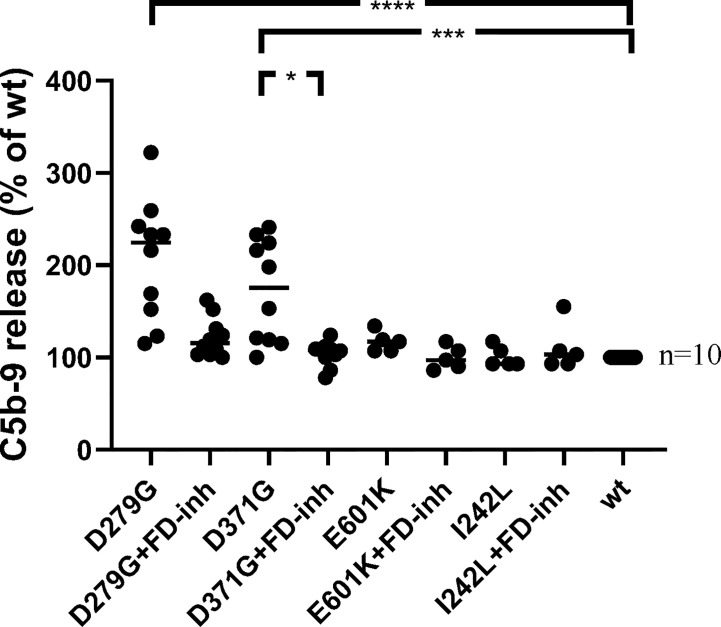
Complement factor B (FB) mutant variants are associated with excessive complement activation in kidney diseases such as atypical hemolytic uremic syndrome (aHUS), C3 glomerulopathy and membranoproliferative glomerulonephritis (MPGN). Patients with aHUS are currently treated with eculizumab while there is no specific treatment for other complement-mediated renal diseases. In this study the phenotype of three FB missense variants, detected in patients with aHUS (D371G and E601K) and MPGN (I242L), was investigated. Patient sera with the D371G and I242L mutations induced hemolysis of sheep erythrocytes. Mutagenesis was performed to study the effect of factor D (FD) inhibition on C3 convertase-induced FB cleavage, complement-mediated hemolysis, and the release of soluble C5b-9 from glomerular endothelial cells. The FD inhibitor danicopan abrogated C3 convertase-associated FB cleavage to the Bb fragment in patient serum, and of the FB constructs, D371G, E601K, I242L, the gain-of-function mutation D279G, and the wild-type construct, in FB-depleted serum. Furthermore, the FD-inhibitor blocked hemolysis induced by the D371G and D279G gain-of-function mutants. In FB-depleted serum the D371G and D279G mutants induced release of C5b-9 from glomerular endothelial cells that was reduced by the FD-inhibitor. These results suggest that FD inhibition can effectively block complement overactivation induced by FB gain-of-function mutations.
Keywords: C3 glomerulopathy; atypical hemolytic uremic syndrome; complement; danicopan; factor B; factor D.
Copyright © 2021 Aradottir, Kristoffersson, Roumenina, Bjerre, Kashioulis, Palsson and Karpman.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, et al. . Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy: Conclusions From a “Kidney Disease: Improving Global Outcomes” (Kdigo) Controversies Conference. Kidney Int (2017) 91:539–51. 10.1016/j.kint.2016.10.005 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous