

Whole Transcriptome Data Analysis Reveals Prognostic Signature Genes for Overall Survival Prediction in Diffuse Large B Cell Lymphoma
- PMID: 34178023
- PMCID: PMC8220154
- DOI: 10.3389/fgene.2021.648800
Whole Transcriptome Data Analysis Reveals Prognostic Signature Genes for Overall Survival Prediction in Diffuse Large B Cell Lymphoma
Abstract
Background: With the improvement of clinical treatment outcomes in diffuse large B cell lymphoma (DLBCL), the high rate of relapse in DLBCL patients is still an established barrier, as the therapeutic strategy selection based on potential targets remains unsatisfactory. Therefore, there is an urgent need in further exploration of prognostic biomarkers so as to improve the prognosis of DLBCL.
Methods: The univariable and multivariable Cox regression models were employed to screen out gene signatures for DLBCL overall survival (OS) prediction. The differential expression analysis was used to identify representative genes in high-risk and low-risk groups, respectively, where student t test and fold change were implemented. The functional difference between the high-risk and low-risk groups was identified by the gene set enrichment analysis.
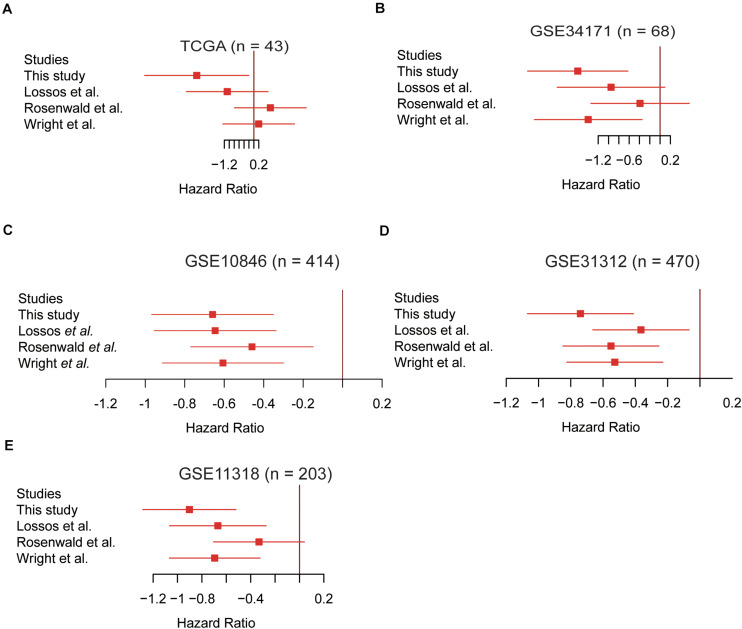
Results: We conducted a systematic data analysis to screen the candidate genes significantly associated with OS of DLBCL in three NCBI Gene Expression Omnibus (GEO) datasets. To construct a prognostic model, five genes (CEBPA, CYP27A1, LST1, MREG, and TARP) were then screened and tested using the multivariable Cox model and the stepwise regression method. Kaplan-Meier curve confirmed the good predictive performance of this five-gene Cox model. Thereafter, the prognostic model and the expression levels of the five genes were validated by means of an independent dataset. High expression levels of these five genes were significantly associated with favorable prognosis in DLBCL, both in training and validation datasets. Additionally, further analysis revealed the independent value and superiority of this prognostic model in risk prediction. Functional enrichment analysis revealed some vital pathways responsible for unfavorable outcome and potential therapeutic targets in DLBCL.
Conclusion: We developed a five-gene Cox model for the clinical outcome prediction of DLBCL patients. Meanwhile, potential drug selection using this model can help clinicians to improve the clinical practice for the benefit of patients.
Keywords: biomarkers; diffuse large B cell lymphoma; overall survival; prognosis; risk score.
Copyright © 2021 Pan, Yang, Wang, Luo, Li, Ding, Lu, Dong, Zhang, Xiu and Liang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





Similar articles
-
Comprehensive analysis of the prognostic implication and immune infiltration of CISD2 in diffuse large B-cell lymphoma.Front Immunol. 2023 Dec 12;14:1277695. doi: 10.3389/fimmu.2023.1277695. eCollection 2023. Front Immunol. 2023. PMID: 38155967 Free PMC article.
-
An integrated prognosis model of pharmacogenomic gene signature and clinical information for diffuse large B-cell lymphoma patients following CHOP-like chemotherapy.J Transl Med. 2020 Mar 30;18(1):144. doi: 10.1186/s12967-020-02311-1. J Transl Med. 2020. PMID: 32228625 Free PMC article.
-
Development of a mitochondria-related gene signature for prognostic assessment in diffuse large B cell lymphoma.Front Oncol. 2025 Mar 20;15:1542829. doi: 10.3389/fonc.2025.1542829. eCollection 2025. Front Oncol. 2025. PMID: 40182032 Free PMC article.
-
Identification and Validation of a Prognostic Prediction Model in Diffuse Large B-Cell Lymphoma.Front Endocrinol (Lausanne). 2022 Apr 14;13:846357. doi: 10.3389/fendo.2022.846357. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35498426 Free PMC article.
-
[Screening of Differential Expression Autophagy Genes Related to the Prognosis of Diffuse Large B-Cell Lymphoma and Establishment of an Autophagy Model].Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2022 Aug;30(4):1101-1108. doi: 10.19746/j.cnki.issn.1009-2137.2022.04.019. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2022. PMID: 35981368 Chinese.
Cited by
-
Genetic and transcriptomic analyses of diffuse large B-cell lymphoma patients with poor outcomes within two years of diagnosis.Leukemia. 2024 Mar;38(3):610-620. doi: 10.1038/s41375-023-02120-7. Epub 2023 Dec 29. Leukemia. 2024. PMID: 38158444 Free PMC article.
-
MicroRNAs in Diffuse Large B-Cell Lymphoma (DLBCL): Biomarkers with Prognostic Potential.Cancers (Basel). 2025 Apr 12;17(8):1300. doi: 10.3390/cancers17081300. Cancers (Basel). 2025. PMID: 40282476 Free PMC article.
-
A novel lipid metabolism-based risk model associated with immunosuppressive mechanisms in diffuse large B-cell lymphoma.Lipids Health Dis. 2024 Jan 22;23(1):20. doi: 10.1186/s12944-024-02017-z. Lipids Health Dis. 2024. PMID: 38254162 Free PMC article.
-
Development and Validation of a Novel Four Gene-Pairs Signature for Predicting Prognosis in DLBCL Patients.Int J Mol Sci. 2024 Nov 28;25(23):12807. doi: 10.3390/ijms252312807. Int J Mol Sci. 2024. PMID: 39684518 Free PMC article.
References
-
- Barrans S. L., Crouch S., Care M. A., Worrillow L., Smith A., Patmore R., et al. (2012). Whole genome expression profiling based on paraffin embedded tissue can be used to classify diffuse large B-cell lymphoma and predict clinical outcome. Br. J. Haematol. 159 441–453. 10.1111/bjh.12045 - DOI - PubMed
LinkOut - more resources
Full Text Sources