

Kmerator Suite: design of specific k-mer signatures and automatic metadata discovery in large RNA-seq datasets
- PMID: 34179780
- PMCID: PMC8221386
- DOI: 10.1093/nargab/lqab058
Kmerator Suite: design of specific k-mer signatures and automatic metadata discovery in large RNA-seq datasets
Abstract
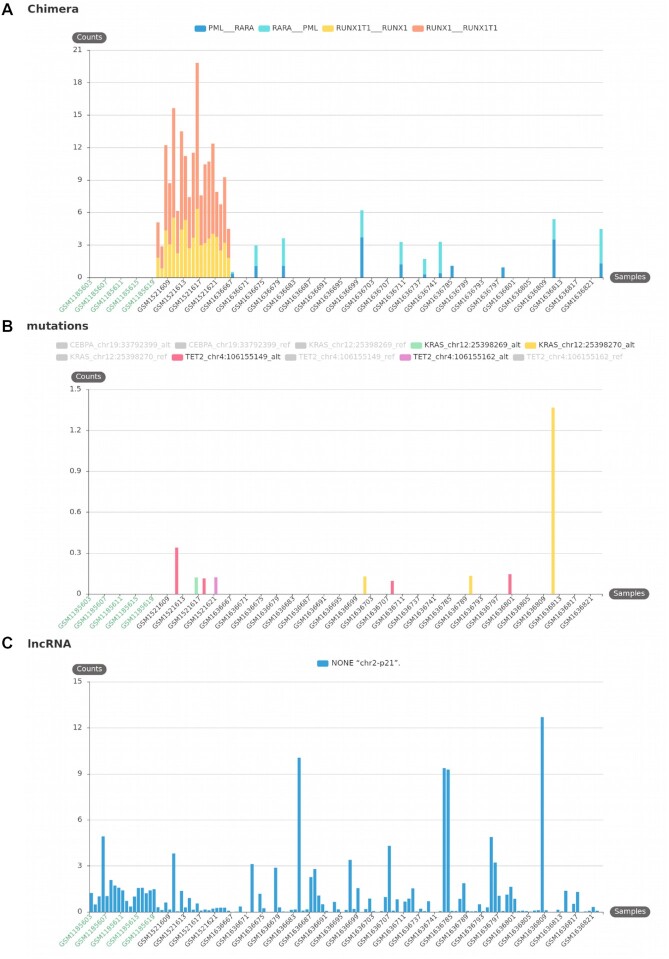
The huge body of publicly available RNA-sequencing (RNA-seq) libraries is a treasure of functional information allowing to quantify the expression of known or novel transcripts in tissues. However, transcript quantification commonly relies on alignment methods requiring a lot of computational resources and processing time, which does not scale easily to large datasets. K-mer decomposition constitutes a new way to process RNA-seq data for the identification of transcriptional signatures, as k-mers can be used to quantify accurately gene expression in a less resource-consuming way. We present the Kmerator Suite, a set of three tools designed to extract specific k-mer signatures, quantify these k-mers into RNA-seq datasets and quickly visualize large dataset characteristics. The core tool, Kmerator, produces specific k-mers for 97% of human genes, enabling the measure of gene expression with high accuracy in simulated datasets. KmerExploR, a direct application of Kmerator, uses a set of predictor gene-specific k-mers to infer metadata including library protocol, sample features or contaminations from RNA-seq datasets. KmerExploR results are visualized through a user-friendly interface. Moreover, we demonstrate that the Kmerator Suite can be used for advanced queries targeting known or new biomarkers such as mutations, gene fusions or long non-coding RNAs for human health applications.
© The Author(s) 2021. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
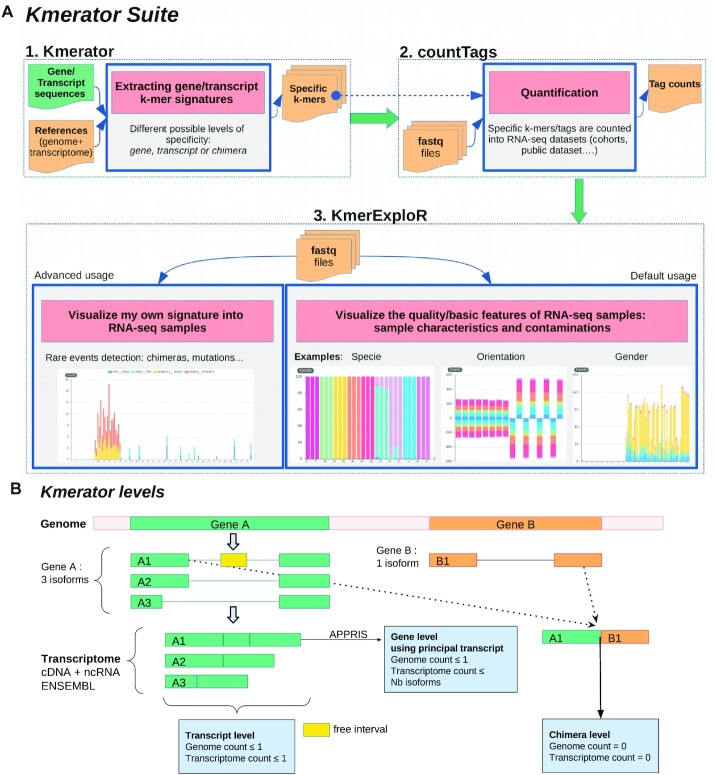
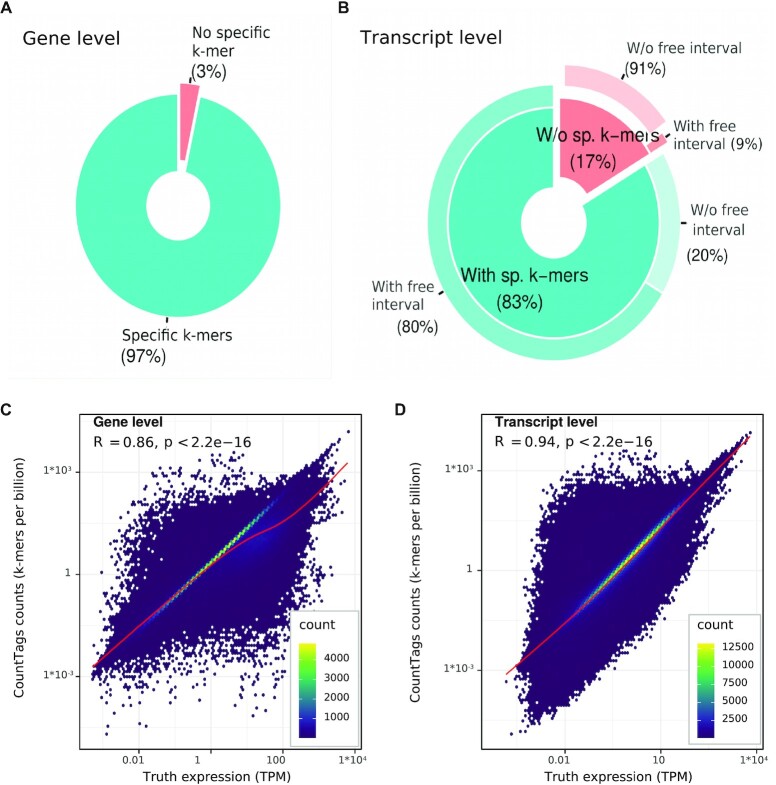
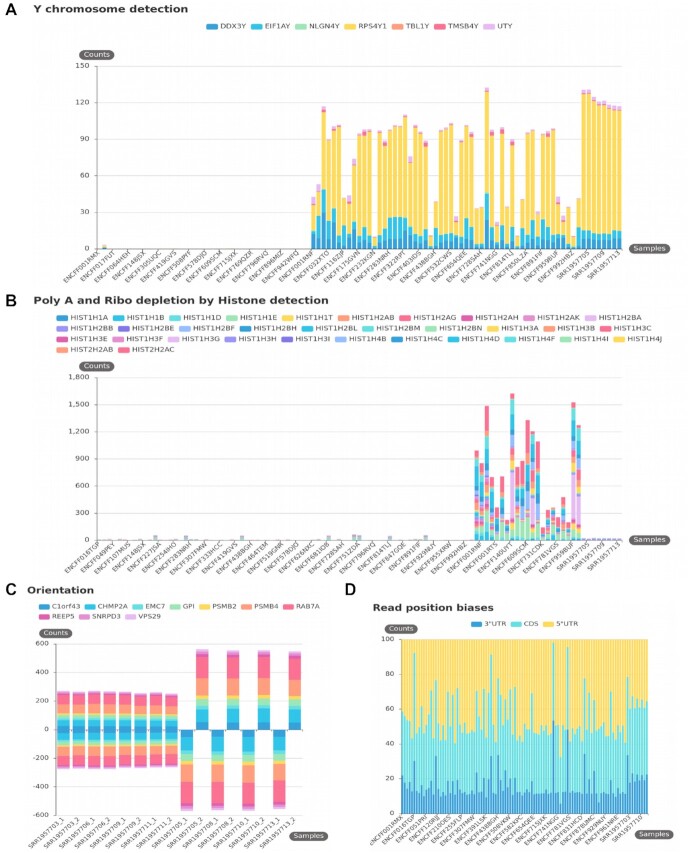
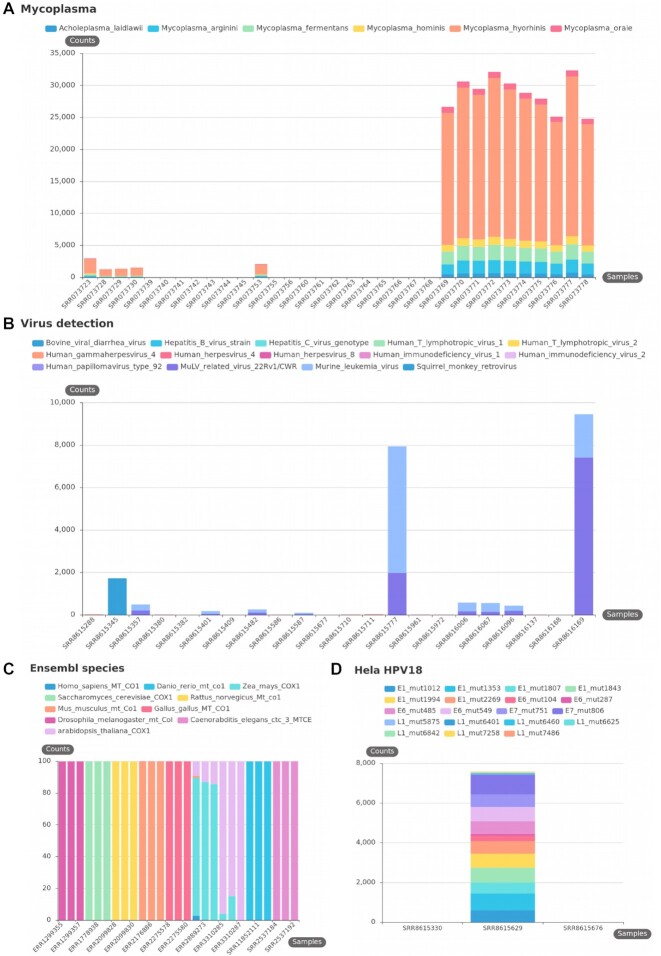
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
