

Exon-Skipping in Duchenne Muscular Dystrophy
- PMID: 34180420
- PMCID: PMC8673534
- DOI: 10.3233/JND-210682
Exon-Skipping in Duchenne Muscular Dystrophy
Abstract
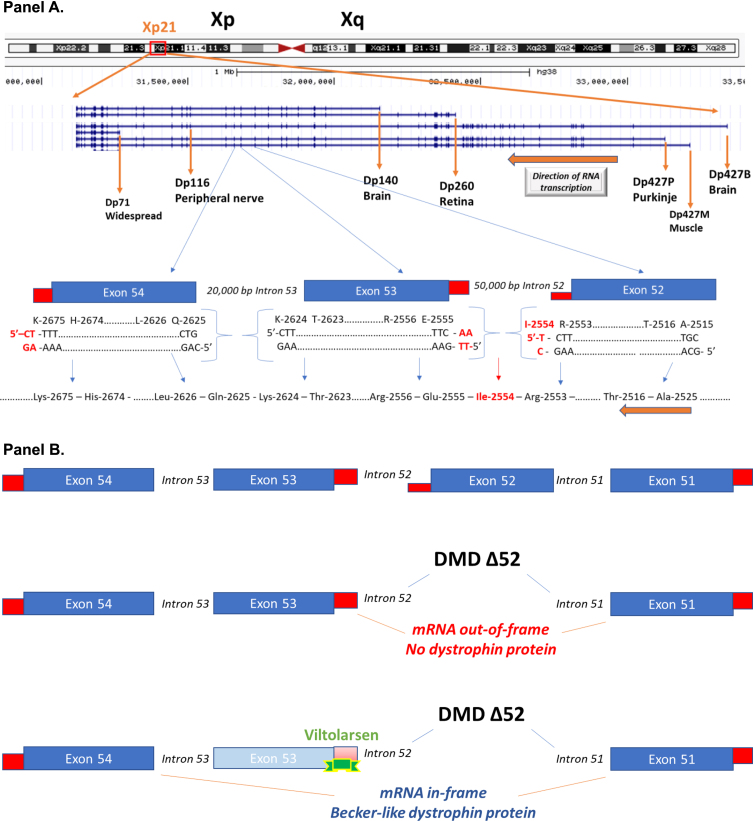
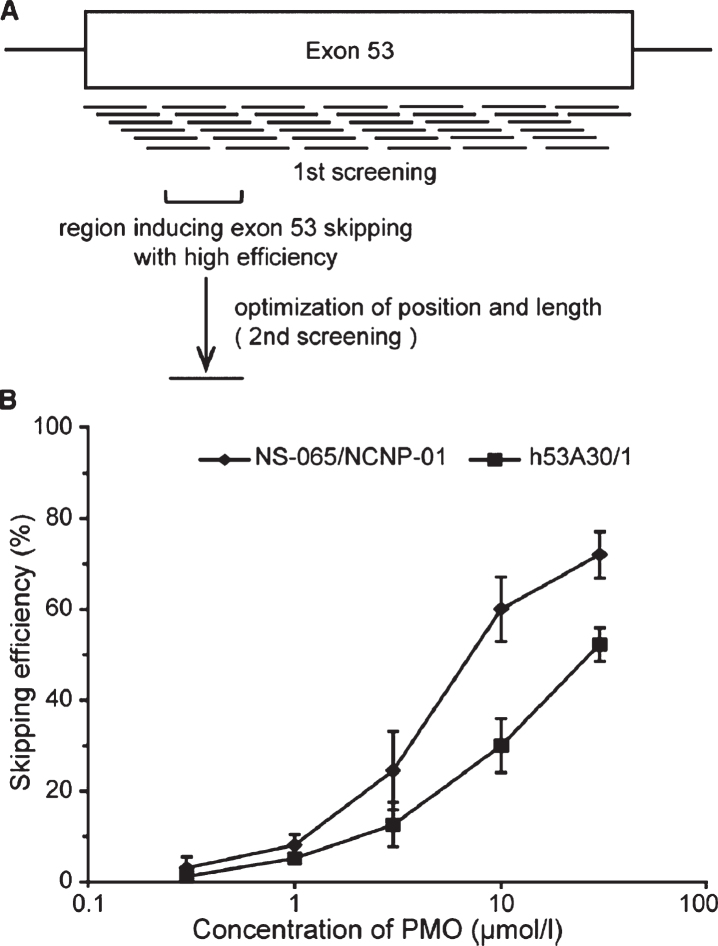
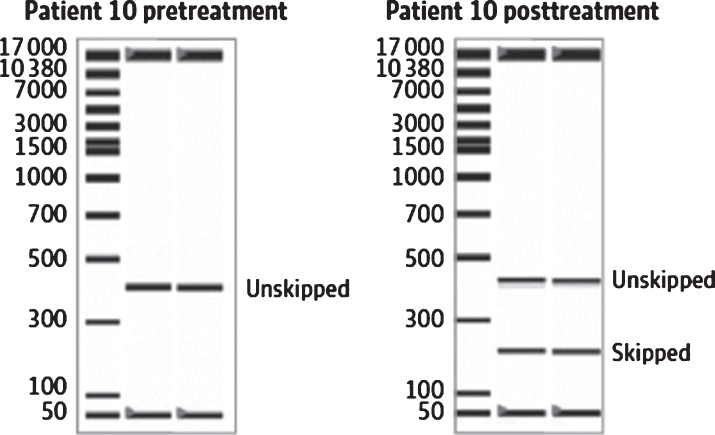
Duchenne muscular dystrophy (DMD) is a devastating, rare disease. While clinically described in the 19th century, the genetic foundation of DMD was not discovered until more than 100 years later. This genetic understanding opened the door to the development of genetic treatments for DMD. Over the course of the last 30 years, the research that supports this development has moved into the realm of clinical trials and regulatory drug approvals. Exon skipping to therapeutically restore the frame of an out-of-frame dystrophin mutation has taken center stage in drug development for DMD. The research reviewed here focuses on the clinical development of exon skipping for the treatment of DMD. In addition to the generation of clinical treatments that are being used for patient care, this research sets the stage for future therapeutic development with a focus on increasing efficacy while providing safety and addressing the multi-systemic aspects of DMD.
Figures
References
-
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–40. - PubMed
-
- Pillers DA, Fitzgerald KM, Duncan NM, Rash SM, White RA, Dwinnell SJ, Powell BR, Schnur RE, Ray PN, Cibis GW, Weleber RG. Duchenne/Becker muscular dystrophy: Correlation of phenotype by electroretinography with sites of dystrophin mutations. Hum Genet. 1999;105(1-2):2–9. doi: 10.1007/s004399900111 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
