

Factors underlying asymmetric pore dynamics of disaggregase and microtubule-severing AAA+ machines
- PMID: 34181904
- PMCID: PMC8391056
- DOI: 10.1016/j.bpj.2021.05.027
Factors underlying asymmetric pore dynamics of disaggregase and microtubule-severing AAA+ machines
Abstract
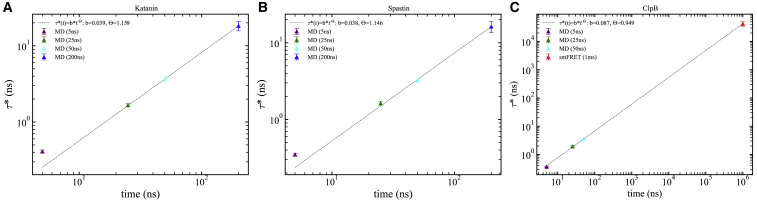
Disaggregation and microtubule-severing nanomachines from the AAA+ (ATPases associated with various cellular activities) superfamily assemble into ring-shaped hexamers that enable protein remodeling by coupling large-scale conformational changes with application of mechanical forces within a central pore by loops protruding within the pore. We probed the asymmetric pore motions and intraring interactions that support them by performing extensive molecular dynamics simulations of single-ring severing proteins and the double-ring disaggregase ClpB. Simulations reveal that dynamic stability of hexameric pores of severing proteins and of the nucleotide-binding domain 1 (NBD1) ring of ClpB, which belong to the same clade, involves a network of salt bridges that connect conserved motifs of central pore loops. Clustering analysis of ClpB highlights correlated motions of domains of neighboring protomers supporting strong interprotomer collaboration. Severing proteins have weaker interprotomer coupling and stronger intraprotomer stabilization through salt bridges involving pore loops. Distinct mechanisms are identified in the NBD2 ring of ClpB involving weaker interprotomer coupling through salt bridges formed by noncanonical loops and stronger intraprotomer coupling. Analysis of collective motions of PL1 loops indicates that the largest amplitude motions in the spiral complex of spastin and ClpB involve axial excursions of the loops, whereas for katanin they involve opening and closing of the central pore. All three motors execute primarily axial excursions in the ring complex. These results suggest distinct substrate processing mechanisms of remodeling and translocation by ClpB and spastin compared to katanin, thus providing dynamic support for the differential action of the two severing proteins. Relaxation dynamics of the distance between the PL1 loops and the center of mass of protomers reveals observation-time-dependent dynamics, leading to predicted relaxation times of tens to hundreds of microseconds on millisecond experimental timescales. For ClpB, the predicted relaxation time is in excellent agreement with the extracted time from smFRET experiments.
Copyright © 2021 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Glover J.R., Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. - PubMed
-
- Erzberger J.P., Berger J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006;35:93–114. - PubMed
-
- Doyle S.M., Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem. Sci. 2009;34:40–48. - PubMed
-
- Lee S., Sowa M.E., Tsai F.T. The ClpB/Hsp104 molecular chaperone-a protein disaggregating machine. J. Struct. Biol. 2004;146:99–105. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
