

Subversion of Lipopolysaccharide Signaling in Gingival Keratinocytes via MCPIP-1 Degradation as a Novel Pathogenic Strategy of Inflammophilic Pathobionts
- PMID: 34182783
- PMCID: PMC8262937
- DOI: 10.1128/mBio.00502-21
Subversion of Lipopolysaccharide Signaling in Gingival Keratinocytes via MCPIP-1 Degradation as a Novel Pathogenic Strategy of Inflammophilic Pathobionts
Abstract
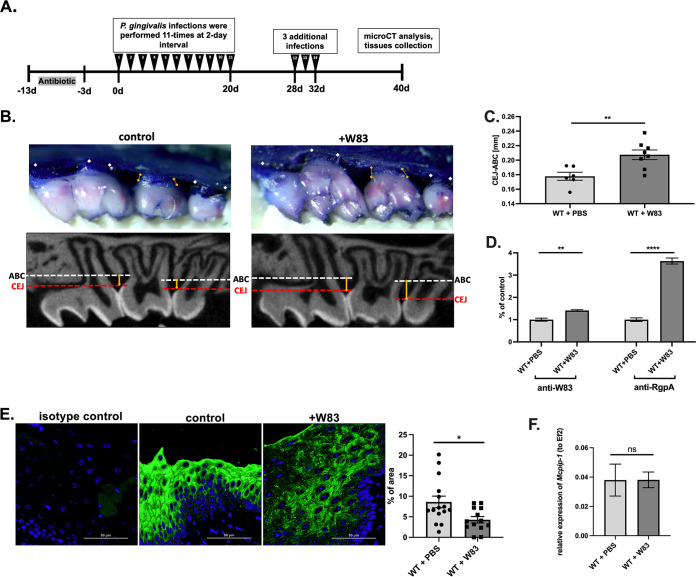
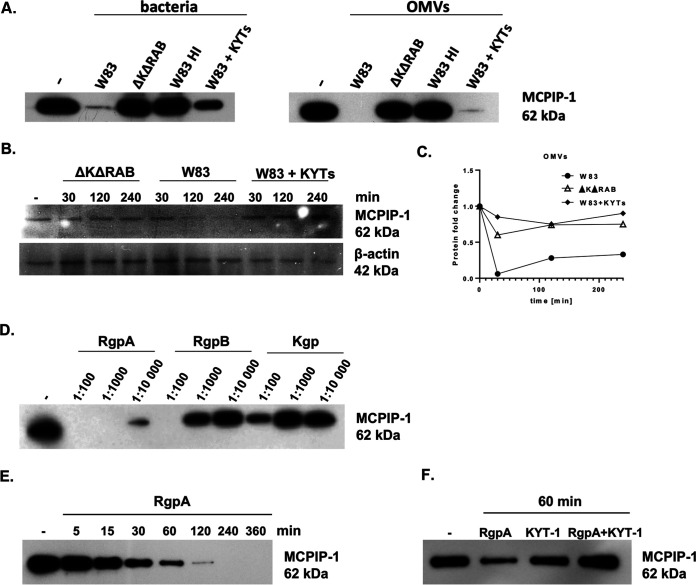
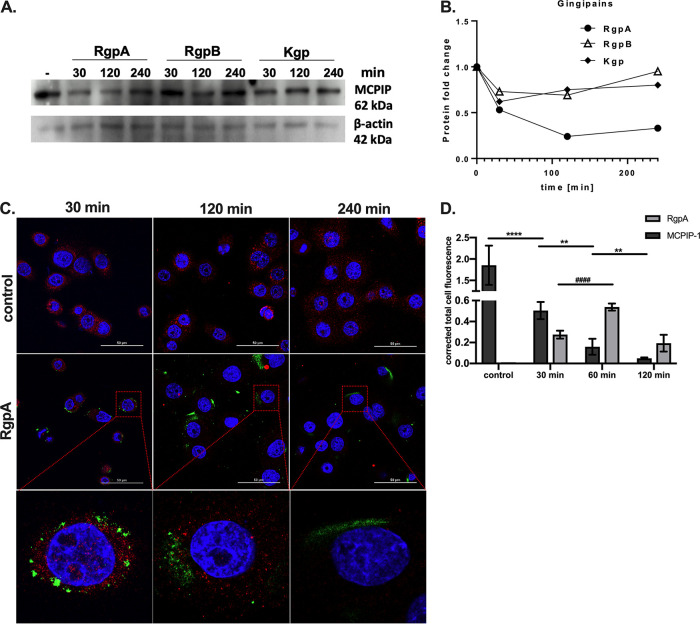
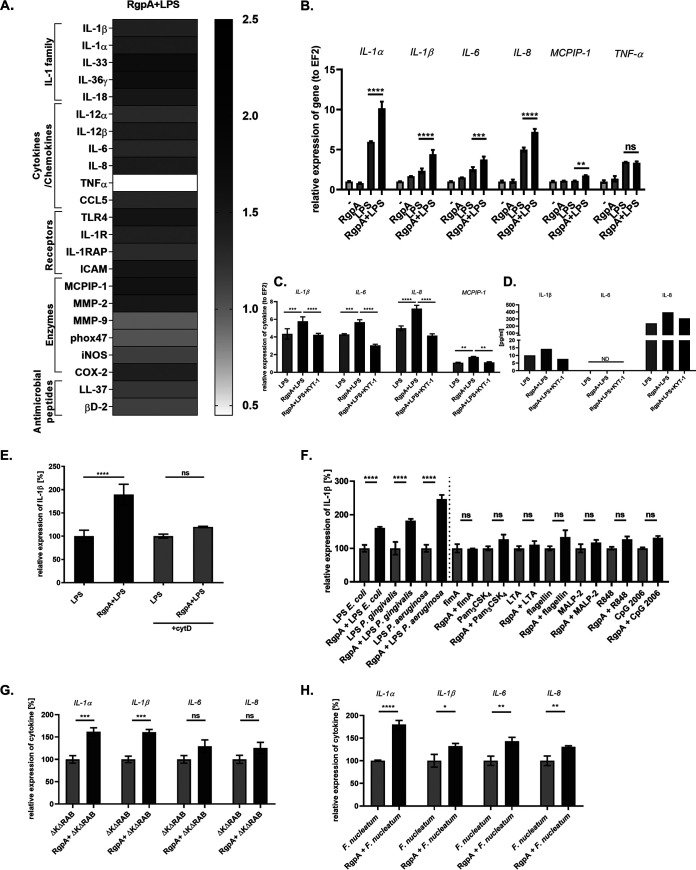
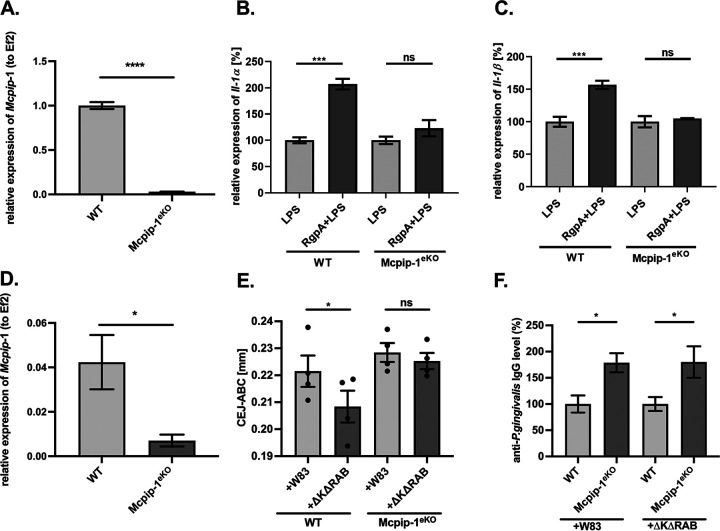
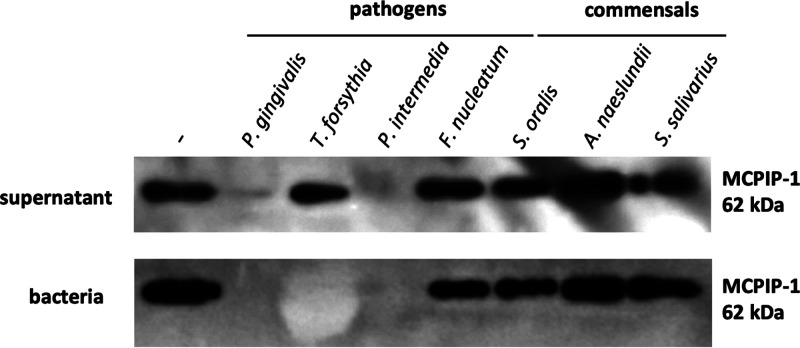
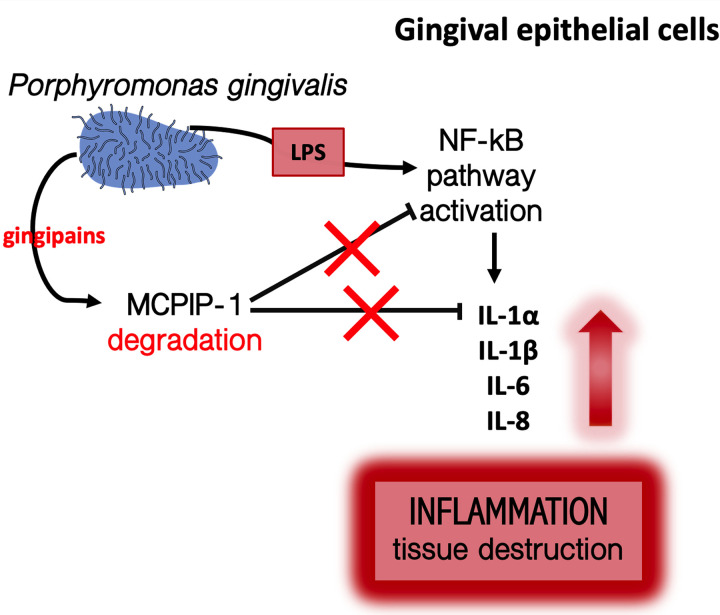
Periodontal disease (PD) is an inflammatory disease of the supporting tissues of the teeth that develops in response to formation of a dysbiotic biofilm on the subgingival tooth surface. Although exacerbated inflammation leads to alveolar bone destruction and may cause tooth loss, the molecular basis of PD initiation and progression remains elusive. Control over the inflammatory reaction and return to homeostasis can be efficiently restored by negative regulators of Toll-like receptor (TLR) signaling pathways such as monocyte chemoattractant protein-induced protein 1 (MCPIP-1), which is constitutively expressed in gingival keratinocytes and prevents hyperresponsiveness in the gingiva. Here, we found that inflammophilic periodontal species influence the stability of MCPIP-1, leading to an aggravated response of the epithelium to proinflammatory stimulation. Among enzymes secreted by periodontal species, gingipains-cysteine proteases from Porphyromonas gingivalis-are considered major contributors to the pathogenic potential of bacteria, strongly influencing the components of the innate and adaptive immune system. Gingipain proteolytic activity leads to a rapid degradation of MCPIP-1, exacerbating the inflammatory response induced by endotoxin. Collectively, these results establish a novel mechanism of corruption of inflammatory signaling by periodontal pathogens, indicating new possibilities for treatment of this chronic disease. IMPORTANCE Periodontitis is a highly prevalent disease caused by accumulation of a bacterial biofilm. Periodontal pathogens use a number of virulence strategies that are under intensive study to find optimal therapeutic approaches against bone loss. In our work, we present a novel mechanism utilized by the key periodontal pathogen Porphyromonas gingivalis, based on the selective degradation of the negative regulator of inflammation, MCPIP-1. We found that the diminished levels of MCPIP-1 in gingival keratinocytes-cells at the forefront of the fight against bacteria-cause sensitization to endotoxins produced by other oral species. This results in an enhanced inflammatory response, which promotes the growth of inflammophilic pathobionts and damage of tooth-supporting tissues. Our observation is relevant to understanding the molecular basis of periodontitis and the development of new methods for treatment.
Keywords: MCPIP-1; Porphyromonas gingivalis; gingipains; lipopolysaccharide; periodontitis.
Figures
Similar articles
-
Regulation of gingival keratinocyte monocyte chemoattractant protein-1-induced protein (MCPIP)-1 and mucosa-associated lymphoid tissue lymphoma translocation protein (MALT)-1 expressions by periodontal bacteria, lipopolysaccharide, and interleukin-1β.J Periodontol. 2023 Jan;94(1):130-140. doi: 10.1002/JPER.22-0093. Epub 2022 Aug 4. J Periodontol. 2023. PMID: 35712915 Free PMC article.
-
Proteolytic Activity-Independent Activation of the Immune Response by Gingipains from Porphyromonas gingivalis.mBio. 2022 Jun 28;13(3):e0378721. doi: 10.1128/mbio.03787-21. Epub 2022 May 2. mBio. 2022. PMID: 35491845 Free PMC article.
-
Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors.Crit Rev Oral Biol Med. 2002;13(2):132-42. doi: 10.1177/154411130201300204. Crit Rev Oral Biol Med. 2002. PMID: 12097356 Review.
-
Localization and expression profiles of gingival monocyte chemoattractant protein-1-induced protein-1 (MCPIP-1) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT-1).Clin Oral Investig. 2023 May;27(5):2065-2074. doi: 10.1007/s00784-023-05010-5. Epub 2023 Apr 3. Clin Oral Investig. 2023. PMID: 37010640 Free PMC article.
-
Pathogenesis of Important Virulence Factors of Porphyromonas gingivalis via Toll-Like Receptors.Front Cell Infect Microbiol. 2019 Jul 18;9:262. doi: 10.3389/fcimb.2019.00262. eCollection 2019. Front Cell Infect Microbiol. 2019. PMID: 31380305 Free PMC article. Review.
Cited by
-
The subversion of toll-like receptor signaling by bacterial and viral proteases during the development of infectious diseases.Mol Aspects Med. 2022 Dec;88:101143. doi: 10.1016/j.mam.2022.101143. Epub 2022 Sep 21. Mol Aspects Med. 2022. PMID: 36152458 Free PMC article. Review.
-
Regulation of gingival keratinocyte monocyte chemoattractant protein-1-induced protein (MCPIP)-1 and mucosa-associated lymphoid tissue lymphoma translocation protein (MALT)-1 expressions by periodontal bacteria, lipopolysaccharide, and interleukin-1β.J Periodontol. 2023 Jan;94(1):130-140. doi: 10.1002/JPER.22-0093. Epub 2022 Aug 4. J Periodontol. 2023. PMID: 35712915 Free PMC article.
-
Periodontal and microbiological data in patients with mucous membrane pemphigoid in a French population in 2021-2022: A pilot cross-sectional study.Health Sci Rep. 2024 Jul 26;7(7):e2163. doi: 10.1002/hsr2.2163. eCollection 2024 Jul. Health Sci Rep. 2024. PMID: 39072352 Free PMC article.
-
Inflammatory macrophages exploited by oral streptococcus increase IL-1B release via NLRP6 inflammasome.J Leukoc Biol. 2023 Sep 27;114(4):347-357. doi: 10.1093/jleuko/qiad089. J Leukoc Biol. 2023. PMID: 37497744 Free PMC article.
-
Proteolytic Activity-Independent Activation of the Immune Response by Gingipains from Porphyromonas gingivalis.mBio. 2022 Jun 28;13(3):e0378721. doi: 10.1128/mbio.03787-21. Epub 2022 May 2. mBio. 2022. PMID: 35491845 Free PMC article.
References
-
- Galimanas V, Hall MW, Singh N, Lynch MDJ, Goldberg M, Tenenbaum H, Cvitkovitch DG, Neufeld JD, Senadheera DB. 2014. Bacterial community composition of chronic periodontitis and novel oral sampling sites for detecting disease indicators. Microbiome 2:32. doi:10.1186/2049-2618-2-32. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
