

Quantifying alternative polyadenylation in RNAseq data with LABRAT
- PMID: 34183124
- PMCID: PMC9041098
- DOI: 10.1016/bs.mie.2021.03.018
Quantifying alternative polyadenylation in RNAseq data with LABRAT
Abstract
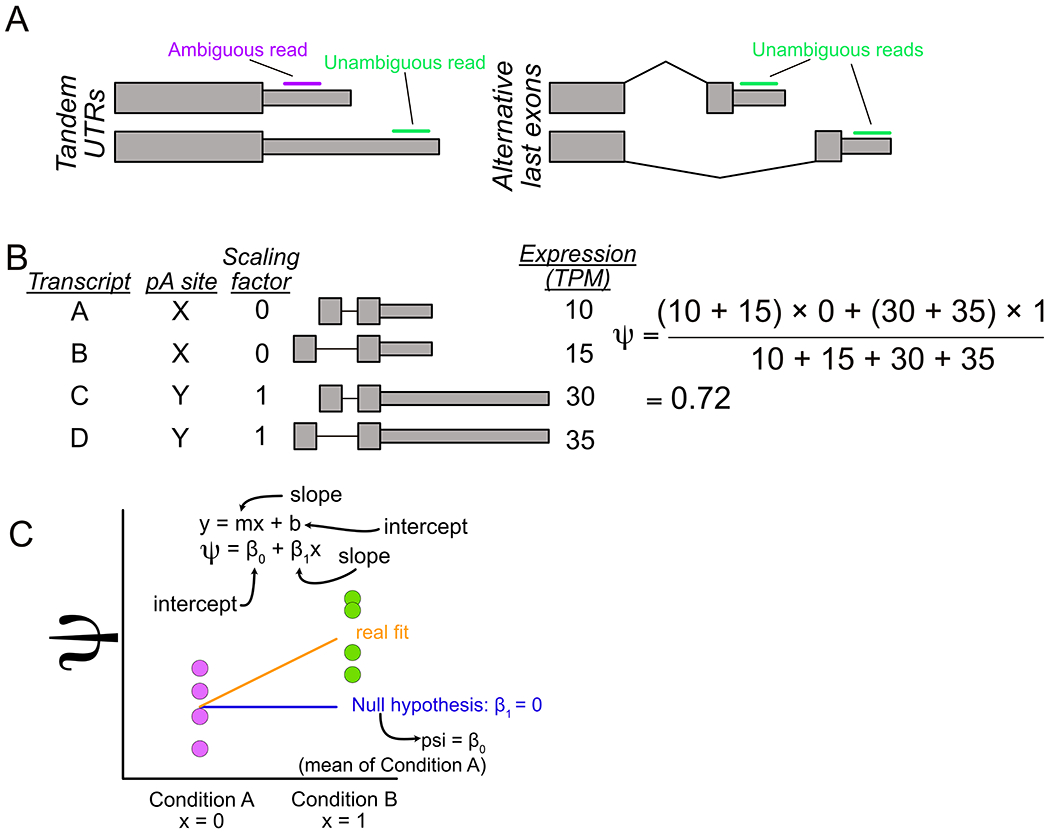
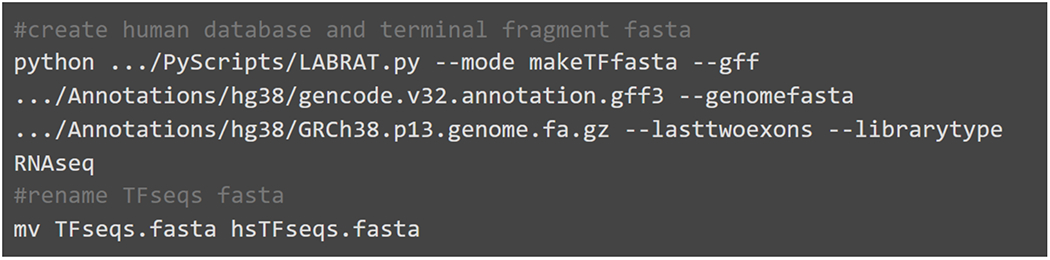
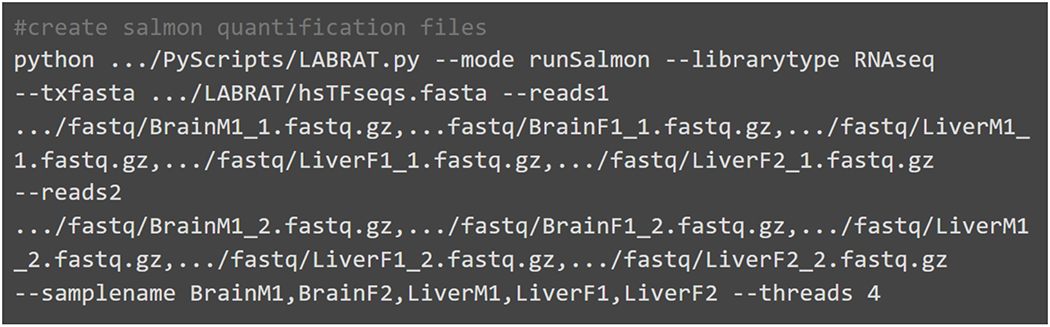
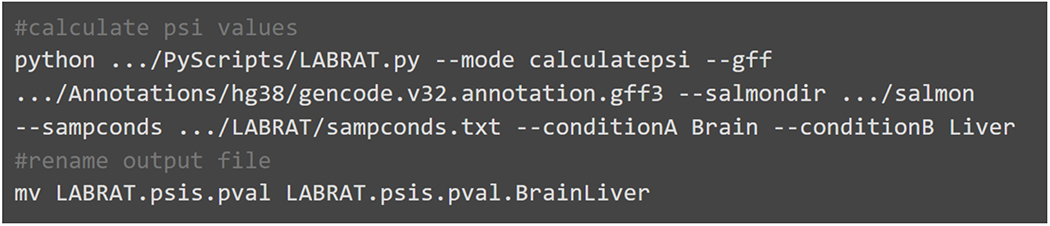
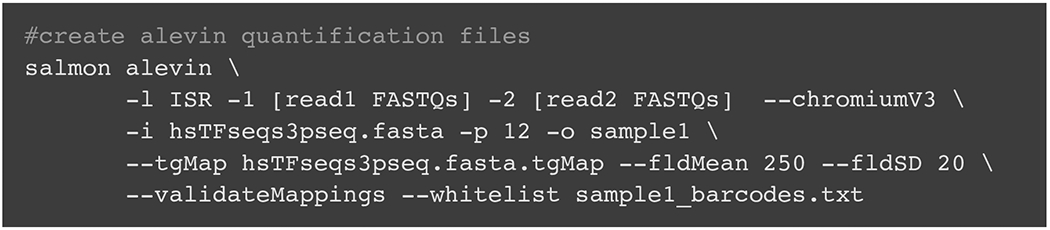
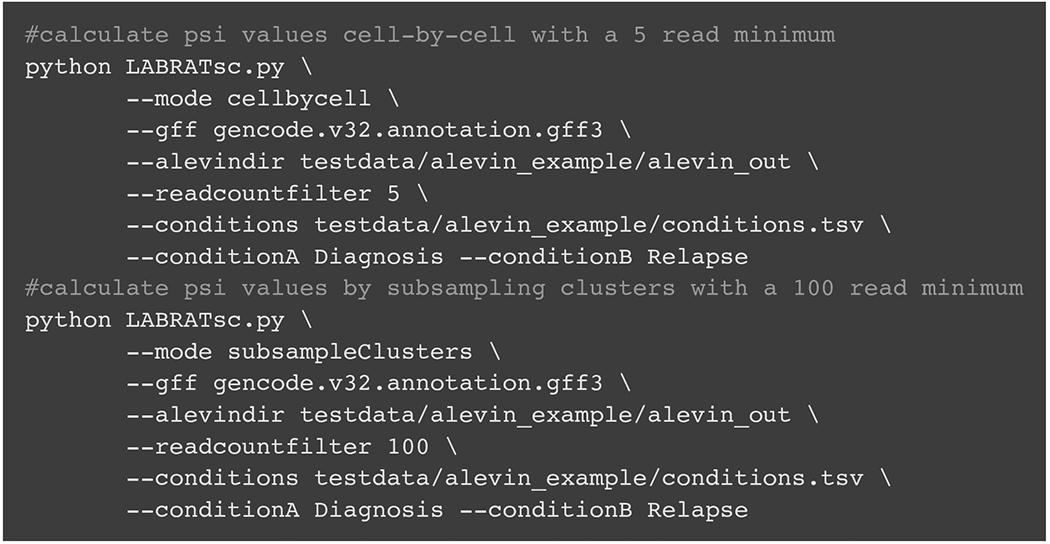
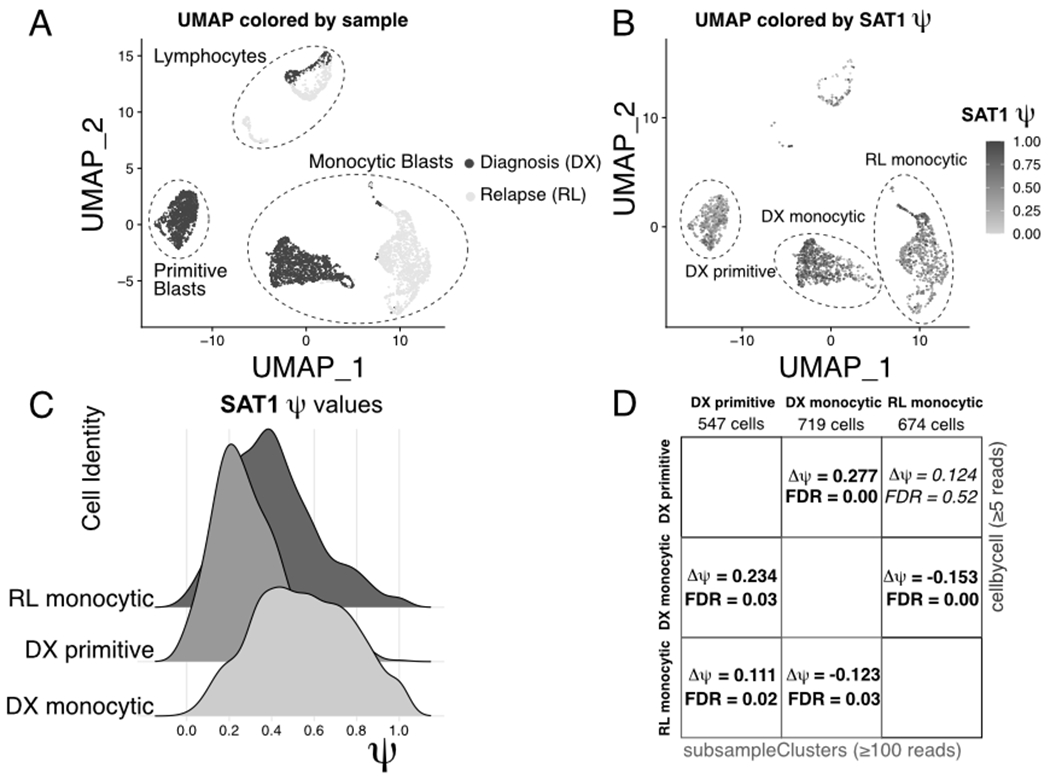
Alternative polyadenylation (APA) generates transcript isoforms that differ in their 3' UTR content and may therefore be subject to different regulatory fates. Although the existence of APA has been known for decades, quantification of APA isoforms from high-throughput RNA sequencing data has been difficult. To facilitate the study of APA in large datasets, we developed an APA quantification technique called LABRAT (Lightweight Alignment-Based Reckoning of Alternative Three-prime ends). LABRAT leverages modern transcriptome quantification approaches to determine the relative abundances of APA isoforms. In this manuscript we describe how LABRAT produces its calculations, provide a step-by-step protocol for its use, and demonstrate its ability to quantify APA in single-cell RNAseq data.
Keywords: 3′ UTR regulation; Alternative polyadenylation; Post-transcriptional regulation; Single cell RNAseq; Transcriptomics.
Copyright © 2021 Elsevier Inc. All rights reserved.
Figures

References
-
- Benjamini Y, & Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B, Statistical Methodology, 57(1), 289–300.
-
- Bray NL, Pimentel H, Melsted P, & Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology, 34(5), 525–527. - PubMed
-
- Cho PF, Poulin F, Cho-Park YA, Cho-Park IB, Chicoine JD, Lasko P, et al. (2005). A new paradigm for translational control: Inhibition via 5′-3’ mRNA tethering by Bicoid and the eIF4E cognate 4EHP. Cell, 121(3), 411–423. - PubMed
-
- Goering R, Engel KL, Gillen AE, Fong N, Bentley DL, & Matthew Taliaferro J (2020). LABRAT reveals association of alternative polyadenylation with transcript localization, RNA binding protein expression, transcription speed, and cancer survival. Cold Spring Harbor Laboratory. 2020.10.05.326702 10.1101/2020.10.05.326702. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
