

Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer
- PMID: 34185431
- PMCID: PMC8181206
- DOI: 10.1002/ctm2.422
Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer
Abstract
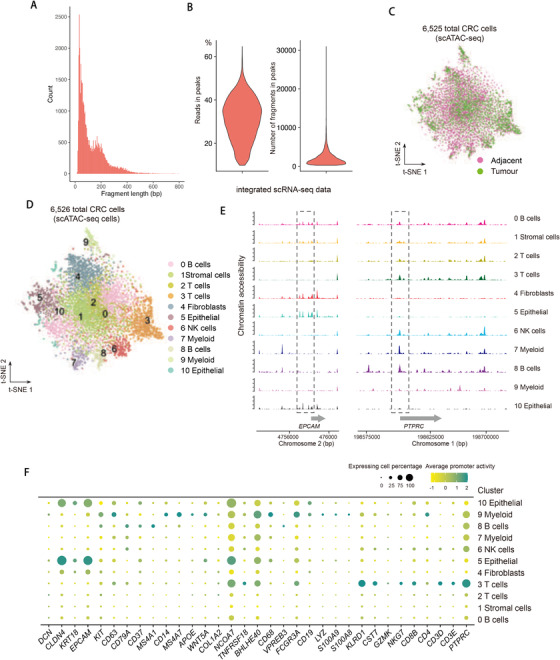
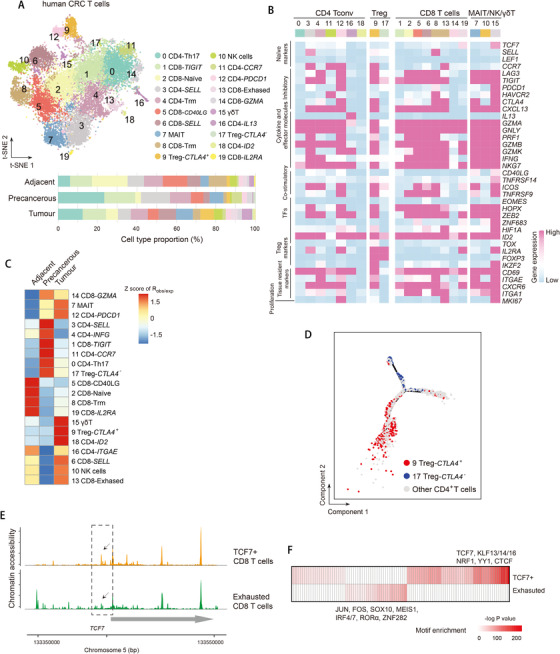
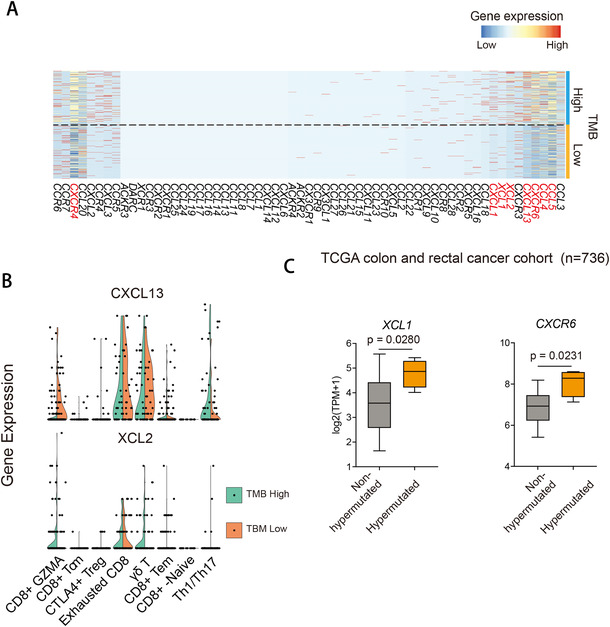
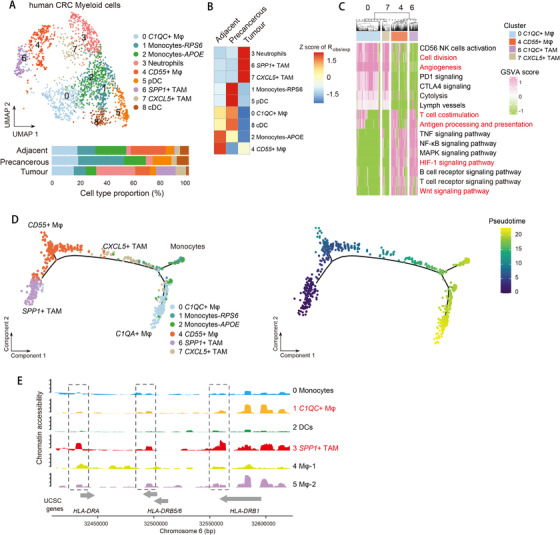
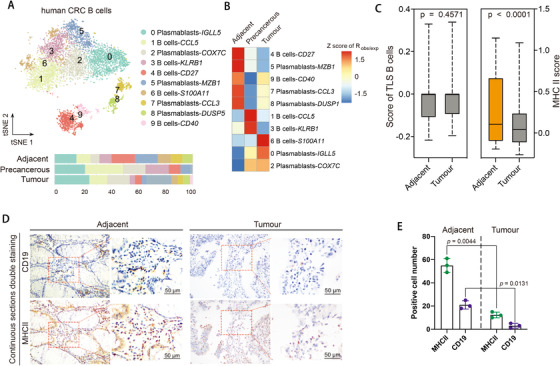
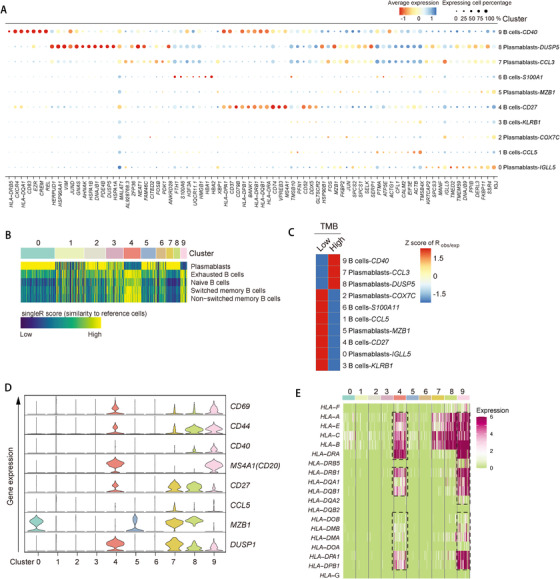
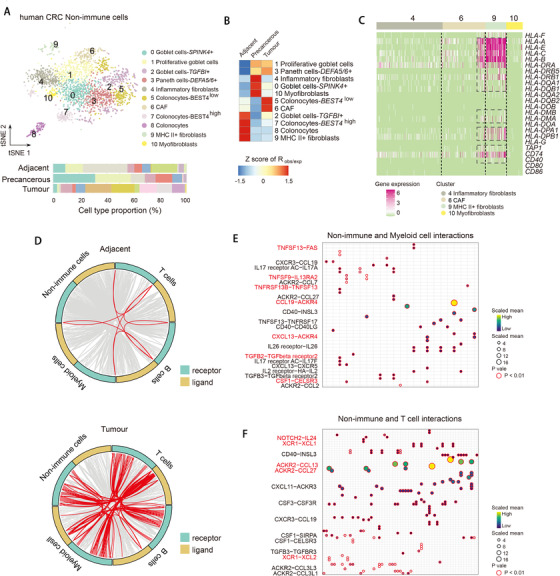
Profiling heterologous cell types within tumors is essential to decipher tumor microenvironment that shapes tumor progress and determines the outcome of therapeutic response. Here, we comprehensively characterized transcriptomes of 34,037 single cells obtained from 12 treatment-naïve patients with colorectal cancer. Our comprehensive evaluation revealed attenuated B-cell antigen presentation, distinct regulatory T-cell clusters with different origin and novel polyfunctional tumor associated macrophages associated with CRC. Moreover, we identified expanded XCL1+ T-cell clusters associated with tumor mutational burden high status. We further explored the underlying molecular mechanisms by profiling epigenetic landscape and inferring transcription factor motifs using single-cell ATAC-seq. Our dataset and analysis approaches herein provide a rich resource for further study of the impact of immune cells and translational research for human colorectal cancer.
Keywords: human colorectal cancer; scATAC-seq; scRNA-seq; tumor immune microenvironment.
© 2021 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
All of the authors declare no interest of conflicts.
Figures

References
-
- Papalexi E, Satija R. Single‐cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2018;18:35‐45. - PubMed
-
- Bian S, Hou Y, Zhou X, et al. Single‐cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362:1060‐1063. - PubMed
-
- Lee H‐O, Hong Y, Etlioglu HE, et al. Lineage‐dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet. 2020;52:594‐603. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical