

Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice
- PMID: 34187891
- PMCID: PMC8271763
- DOI: 10.1073/pnas.2100356118
Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice
Abstract
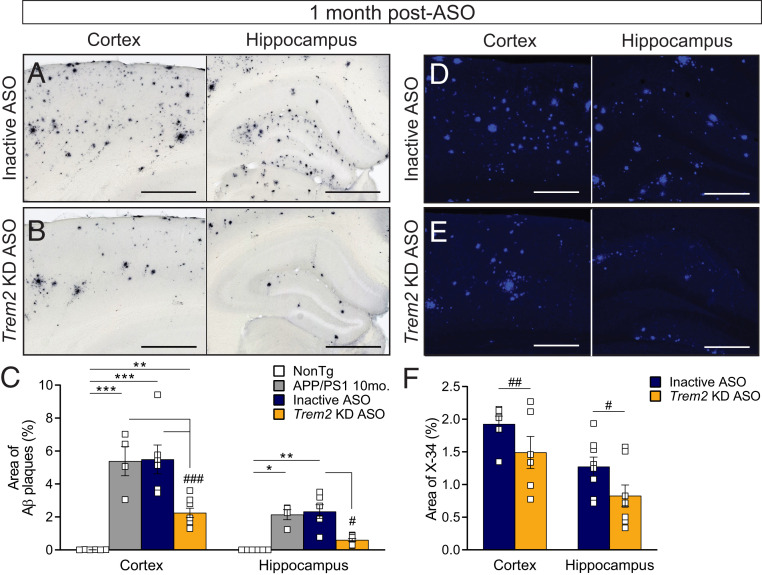
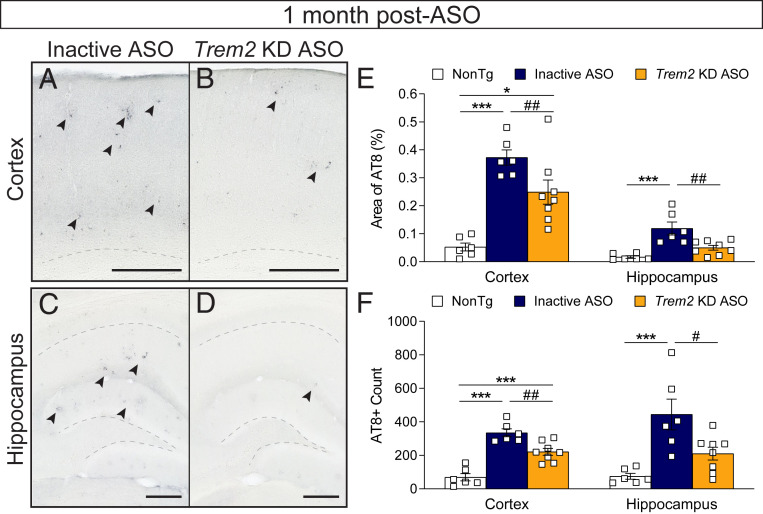
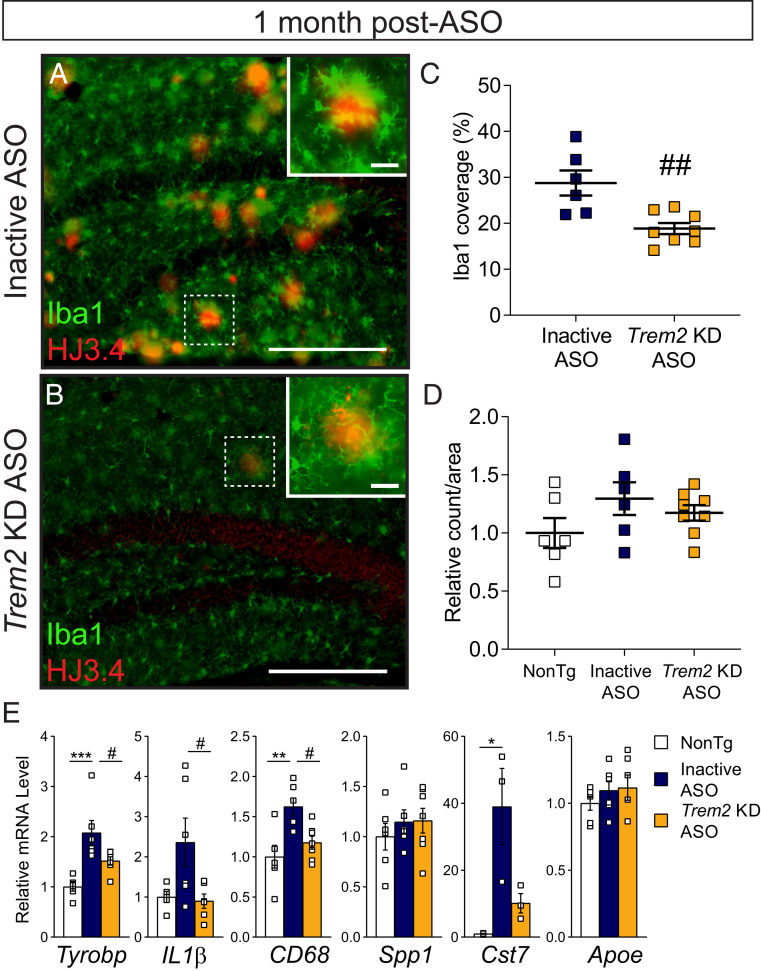
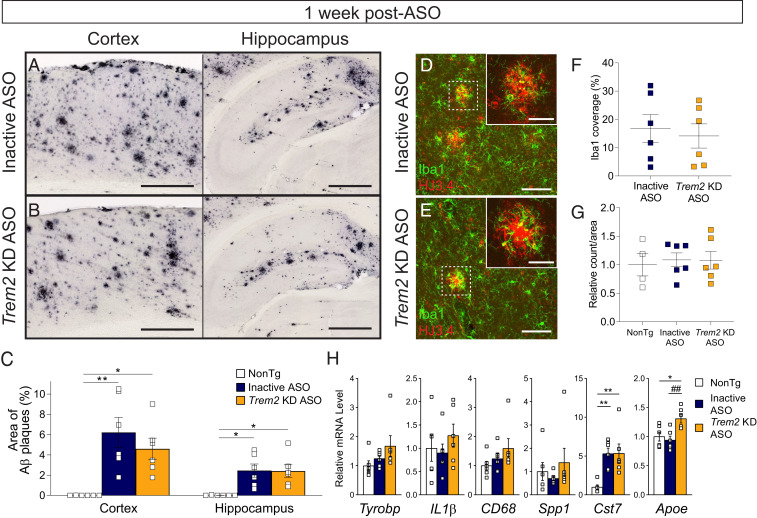
Heterozygous genetic variants within the TREM2 gene show a strong association with increased Alzheimer's disease (AD) risk. Amyloid beta-depositing mouse models haploinsufficient or null for Trem2 have identified important relationships among TREM2, microglia, and AD pathology; however, results are challenging to interpret in the context of varying microglial phenotypes and disease progression. We hypothesized that acute Trem2 reduction may alter amyloid pathology and microglial responses independent of genetic Trem2 deletion in mouse models. We developed antisense oligonucleotides (ASOs) that potently but transiently lower Trem2 messenger RNA throughout the brain and administered them to APP/PS1 mice at varying stages of plaque pathology. Late-stage ASO-mediated Trem2 knockdown significantly reduced plaque deposition and attenuated microglial association around plaque deposits when evaluated 1 mo after ASO injection. Changes in microglial gene signatures 1 wk after ASO administration and phagocytosis measured in ASO-treated cells together indicate that microglia may be activated with short-term Trem2 reduction. These results suggest a time- and/or dose-dependent role for TREM2 in mediating plaque deposition and microglial responses in which loss of TREM2 function may be beneficial for microglial activation and plaque removal in an acute context.
Keywords: Alzheimer’s disease; Trem2; amyloid; antisense oligonucleotide; microglia.
Conflict of interest statement
Competing interest statement: P.J.-N. and F.R. are paid employees of Ionis Pharmaceuticals. Washington University in St. Louis has an existing licensing agreement with Ionis Pharmaceuticals for use of antisense oligonucleotides in these studies. T.M.M. is a consultant for Ionis Pharmaceuticals.
Figures


References
-
- Calsolaro V., Edison P., Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 12, 719–732 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
