Pangenomics reveals alternative environmental lifestyles among chlamydiae
- PMID: 34188040
- PMCID: PMC8242063
- DOI: 10.1038/s41467-021-24294-3
Pangenomics reveals alternative environmental lifestyles among chlamydiae
Abstract
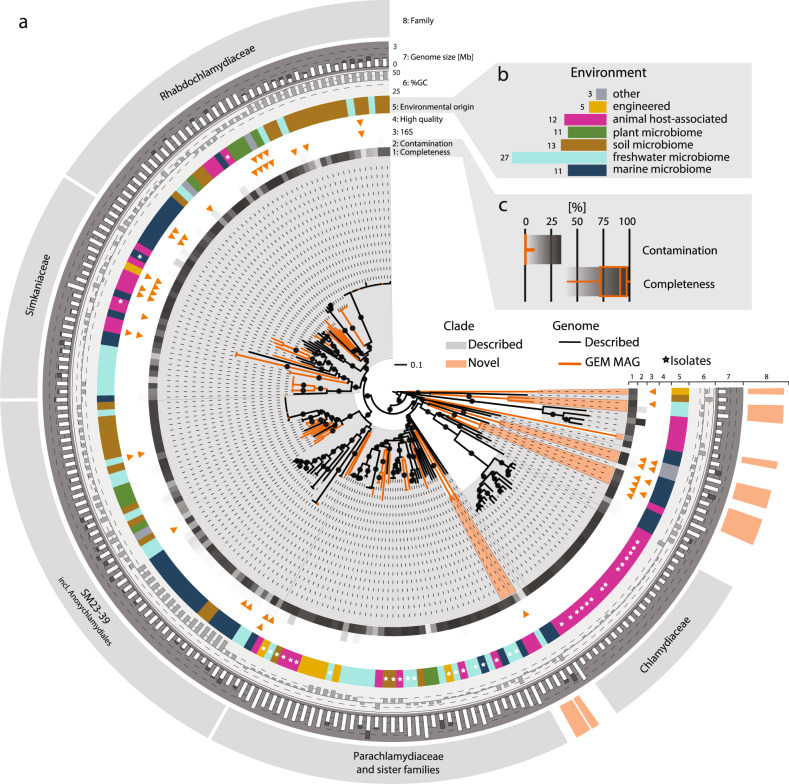
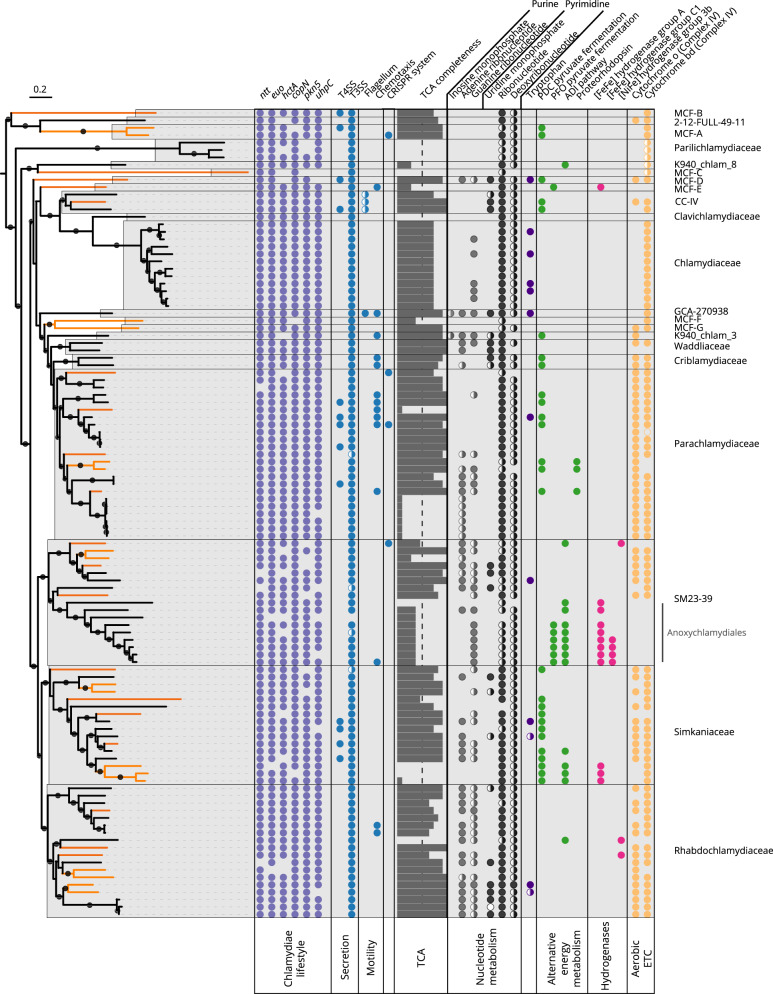
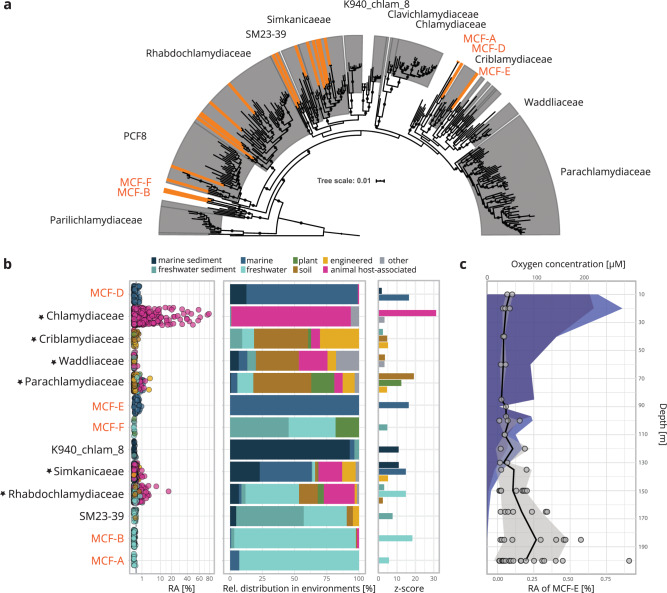
Chlamydiae are highly successful strictly intracellular bacteria associated with diverse eukaryotic hosts. Here we analyzed metagenome-assembled genomes of the "Genomes from Earth's Microbiomes" initiative from diverse environmental samples, which almost double the known phylogenetic diversity of the phylum and facilitate a highly resolved view at the chlamydial pangenome. Chlamydiae are defined by a relatively large core genome indicative of an intracellular lifestyle, and a highly dynamic accessory genome of environmental lineages. We observe chlamydial lineages that encode enzymes of the reductive tricarboxylic acid cycle and for light-driven ATP synthesis. We show a widespread potential for anaerobic energy generation through pyruvate fermentation or the arginine deiminase pathway, and we add lineages capable of molecular hydrogen production. Genome-informed analysis of environmental distribution revealed lineage-specific niches and a high abundance of chlamydiae in some habitats. Together, our data provide an extended perspective of the variability of chlamydial biology and the ecology of this phylum of intracellular microbes.
Conflict of interest statement
The authors declare no competing interests.
Figures