

This is a preprint.
SARS-CoV-2 under an elimination strategy in Hong Kong
- PMID: 34189537
- PMCID: PMC8240692
- DOI: 10.1101/2021.06.19.21259169
SARS-CoV-2 under an elimination strategy in Hong Kong
Update in
-
Genomic epidemiology of SARS-CoV-2 under an elimination strategy in Hong Kong.Nat Commun. 2022 Feb 8;13(1):736. doi: 10.1038/s41467-022-28420-7. Nat Commun. 2022. PMID: 35136039 Free PMC article.
Abstract
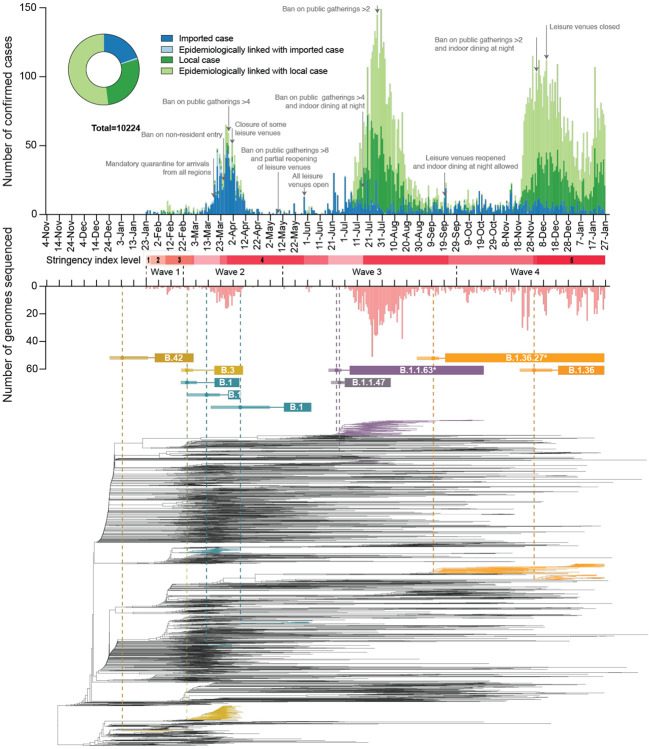
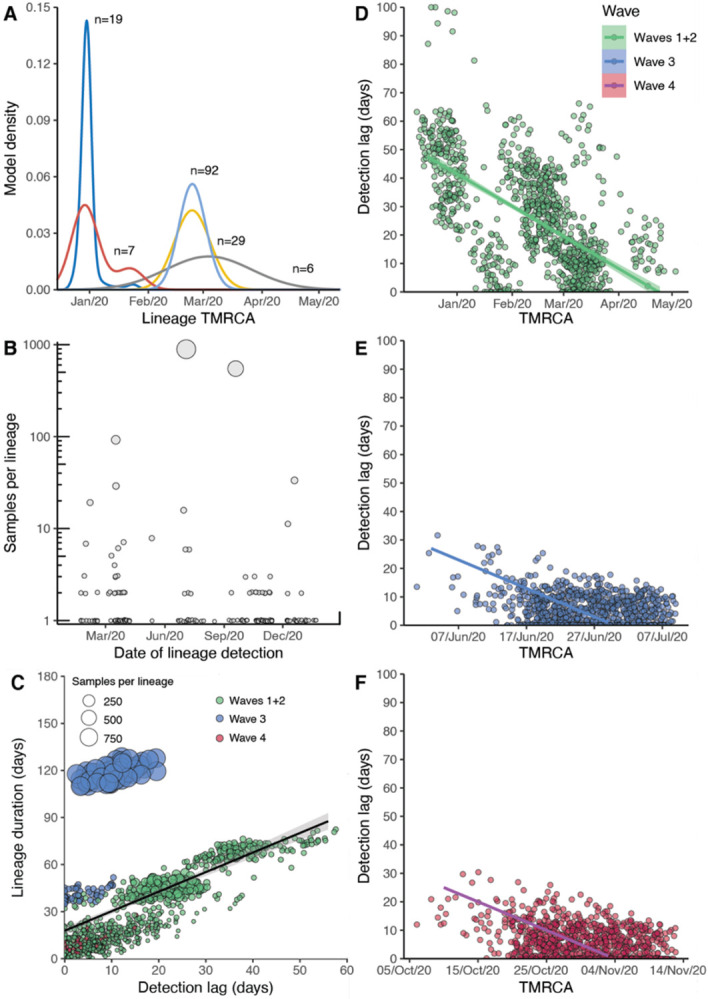
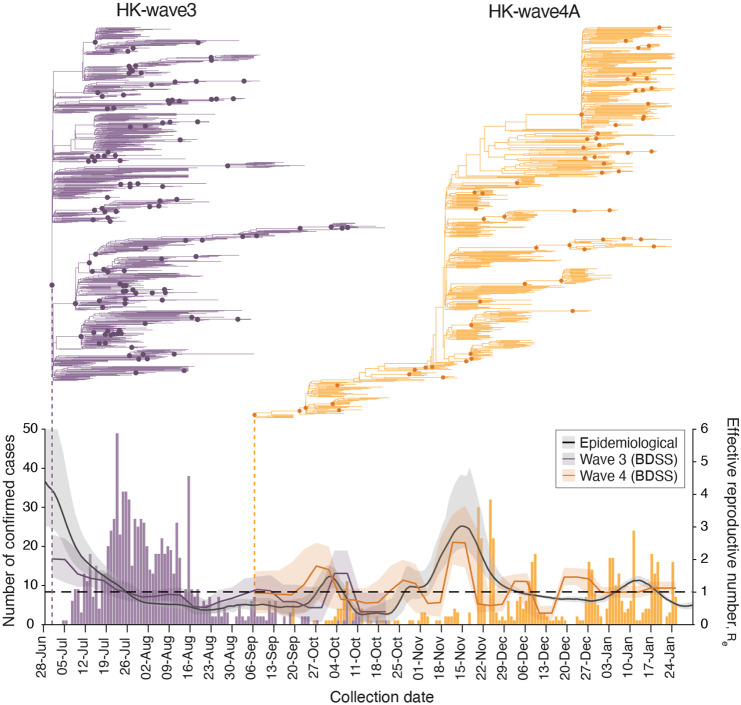
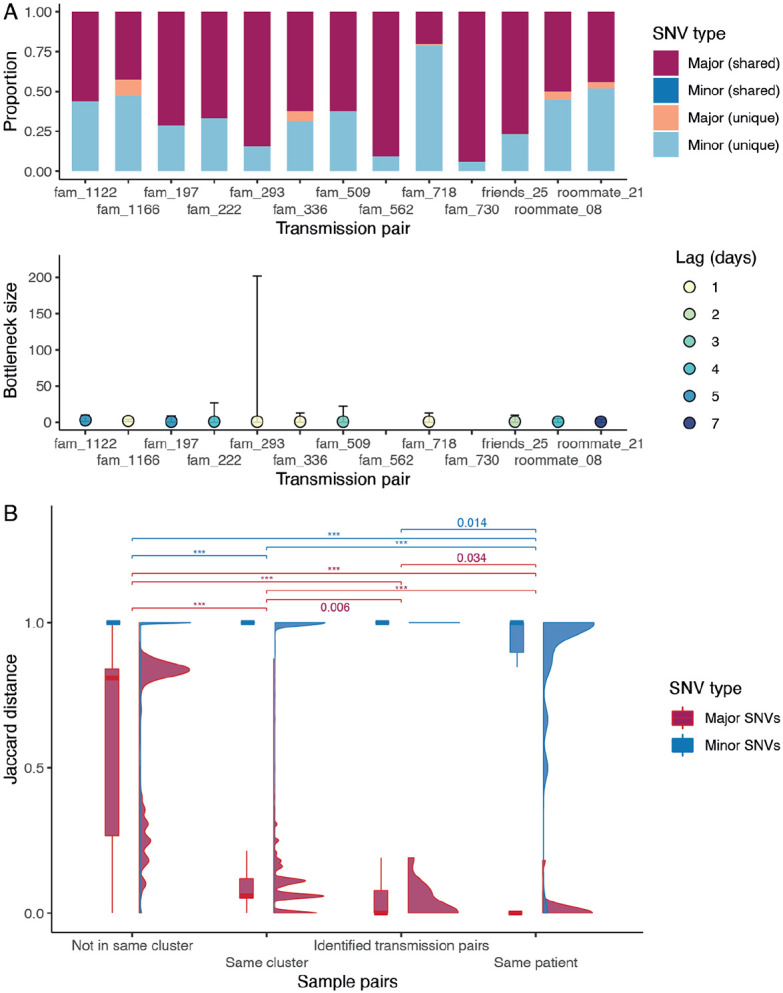
Hong Kong utilized an elimination strategy with intermittent use of public health and social measures and increasingly stringent travel regulations to control SARS-CoV-2 transmission. By analyzing >1700 genome sequences representing 17% of confirmed cases from 23-January-2020 to 26-January-2021, we reveal the effects of fluctuating control measures on the evolution and epidemiology of SARS-CoV-2 lineages in Hong Kong. Despite numerous importations, only three introductions were responsible for 90% of locally-acquired cases, two of which circulated cryptically for weeks while less stringent measures were in place. We found that SARS-CoV-2 within-host diversity was most similar among transmission pairs and epidemiological clusters due to a strong transmission bottleneck through which similar genetic background generates similar within-host diversity.
One sentence summary: Out of the 170 detected introductions of SARS-CoV-2 in Hong Kong during 2020, three introductions caused 90% of community cases.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous