

Complement mediates binding and procoagulant effects of ultralarge HIT immune complexes
- PMID: 34189574
- PMCID: PMC8617432
- DOI: 10.1182/blood.2020009487
Complement mediates binding and procoagulant effects of ultralarge HIT immune complexes
Abstract
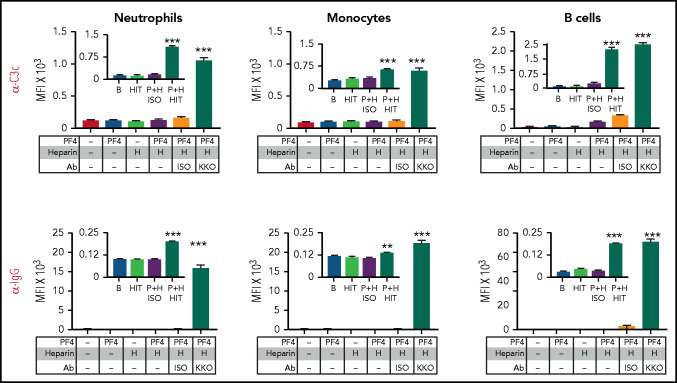
Heparin-induced thrombocytopenia (HIT) is a prothrombotic disorder mediated by ultra-large immune complexes (ULICs) containing immunoglobulin G (IgG) antibodies to a multivalent antigen composed of platelet factor 4 and heparin. The limitations of current antithrombotic therapy in HIT supports the need to identify additional pathways that may be targets for therapy. Activation of FcγRIIA by HIT ULICs initiates diverse procoagulant cellular effector functions. HIT ULICs are also known to activate complement, but the contribution of this pathway to the pathogenesis of HIT has not been studied in detail. We observed that HIT ULICs physically interact with C1q in buffer and plasma, activate complement via the classical pathway, promote codeposition of IgG and C3 complement fragments (C3c) on neutrophil and monocyte cell surfaces. Complement activation by ULICs, in turn, facilitates FcγR-independent monocyte tissue factor expression, enhances IgG binding to the cell surface FcγRs, and promotes platelet adhesion to injured endothelium. Inhibition of the proximal, but not terminal, steps in the complement pathway abrogates monocyte tissue factor expression by HIT ULICs. Together, these studies suggest a major role for complement activation in regulating Fc-dependent effector functions of HIT ULICs, identify potential non-anticoagulant targets for therapy, and provide insights into the broader roles of complement in immune complex-mediated thrombotic disorders.
© 2021 by The American Society of Hematology.
Figures







Similar articles
-
Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia.Blood. 2016 Jan 28;127(4):464-72. doi: 10.1182/blood-2013-11-539262. Epub 2015 Oct 30. Blood. 2016. PMID: 26518435 Free PMC article.
-
Modulation of ultralarge immune complexes in heparin-induced thrombocytopenia.J Thromb Haemost. 2023 Mar;21(3):652-666. doi: 10.1016/j.jtha.2022.11.043. Epub 2022 Dec 22. J Thromb Haemost. 2023. PMID: 36696211
-
Destabilization of PF4-antigenic complexes in heparin-induced thrombocytopenia.Blood. 2025 Jun 19;145(25):3030-3040. doi: 10.1182/blood.2024025653. Blood. 2025. PMID: 40132149
-
Heparin-induced thrombocytopenia: a ten-year retrospective.Annu Rev Med. 1999;50:129-47. doi: 10.1146/annurev.med.50.1.129. Annu Rev Med. 1999. PMID: 10073268 Review.
-
Immunobiology of heparin-induced thrombocytopenia.Curr Top Microbiol Immunol. 2010;341:193-202. doi: 10.1007/82_2010_17. Curr Top Microbiol Immunol. 2010. PMID: 20369319 Review.
Cited by
-
Prothrombotic Phenotype in COVID-19: Focus on Platelets.Int J Mol Sci. 2021 Dec 20;22(24):13638. doi: 10.3390/ijms222413638. Int J Mol Sci. 2021. PMID: 34948438 Free PMC article. Review.
-
Live imaging of platelets and neutrophils during antibody-mediated neurovascular thrombosis.Blood Adv. 2022 Jun 28;6(12):3697-3702. doi: 10.1182/bloodadvances.2021006728. Blood Adv. 2022. PMID: 35452514 Free PMC article.
-
Thrombotic anti-PF4 immune disorders: HIT, VITT, and beyond.Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):1-10. doi: 10.1182/hematology.2023000503. Hematology Am Soc Hematol Educ Program. 2023. PMID: 38066843 Free PMC article.
-
Endothelial cell activation enhances thromboinflammation in vaccine-induced immune thrombotic thrombocytopenia.Blood Adv. 2025 Jun 24;9(12):2891-2906. doi: 10.1182/bloodadvances.2024014165. Blood Adv. 2025. PMID: 40085945 Free PMC article.
-
Platelet phosphatidylserine is the critical mediator of thrombosis in heparin-induced thrombocytopenia.Haematologica. 2023 Oct 1;108(10):2690-2702. doi: 10.3324/haematol.2022.282275. Haematologica. 2023. PMID: 37102605 Free PMC article.
References
-
- Reilly MP, Taylor SM, Hartman NK, et al. . Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcgammaRIIA. Blood. 2001;98(8):2442-2447. - PubMed
-
- Lee DH, Warkentin TE, Denomme GA, Hayward CP, Kelton JG. A diagnostic test for heparin-induced thrombocytopenia: detection of platelet microparticles using flow cytometry. Br J Haematol. 1996; 95(4):724-731. - PubMed
-
- Hughes M, Hayward CP, Warkentin TE, Horsewood P, Chorneyko KA, Kelton JG. Morphological analysis of microparticle generation in heparin-induced thrombocytopenia. Blood. 2000;96(1):188-194. - PubMed
-
- Pouplard C, Iochmann S, Renard B, et al. . Induction of monocyte tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood. 2001; 97(10):3300-3302. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
