

Hepatocyte membrane potential regulates serum insulin and insulin sensitivity by altering hepatic GABA release
- PMID: 34192533
- PMCID: PMC8341405
- DOI: 10.1016/j.celrep.2021.109298
Hepatocyte membrane potential regulates serum insulin and insulin sensitivity by altering hepatic GABA release
Abstract
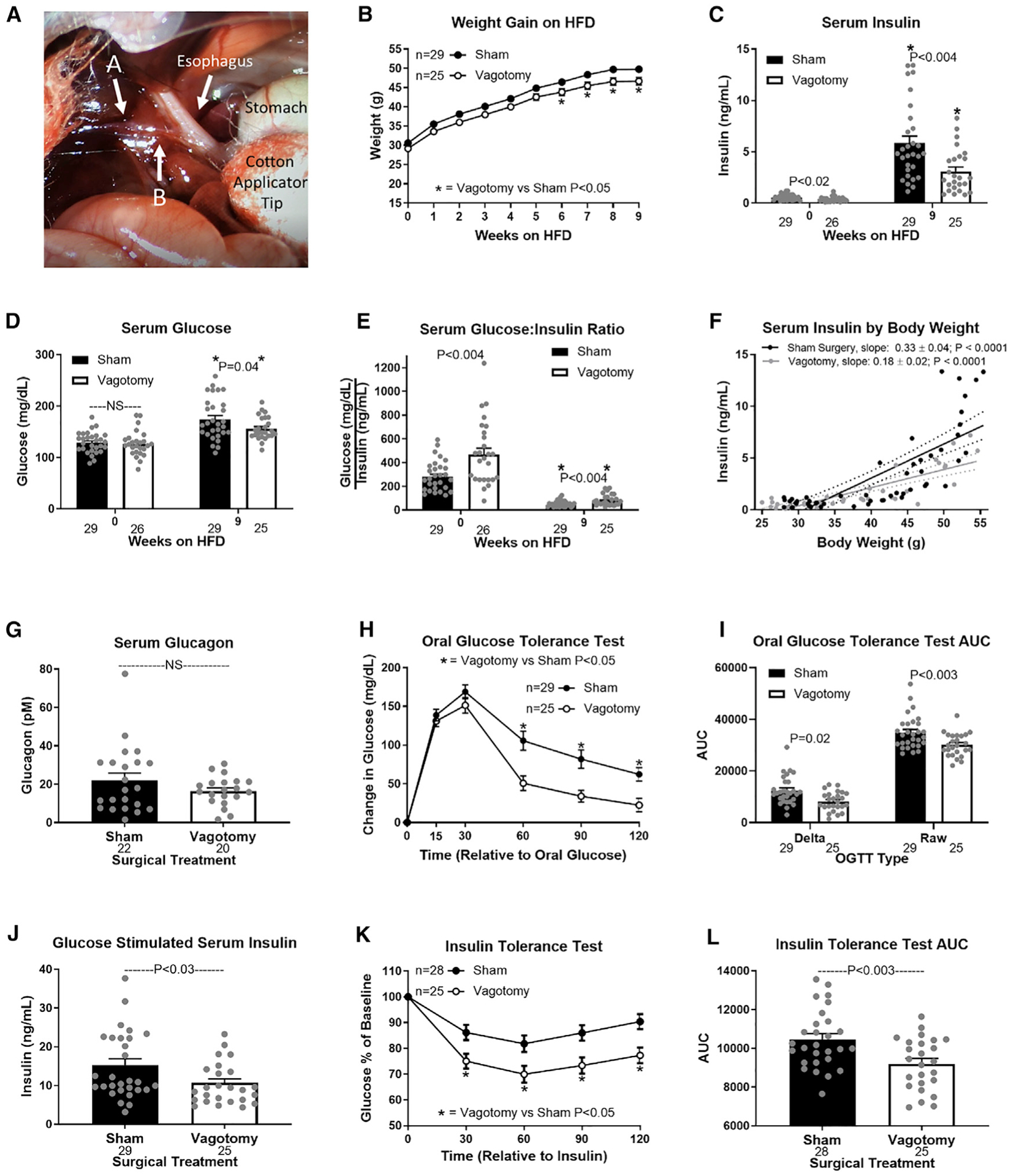
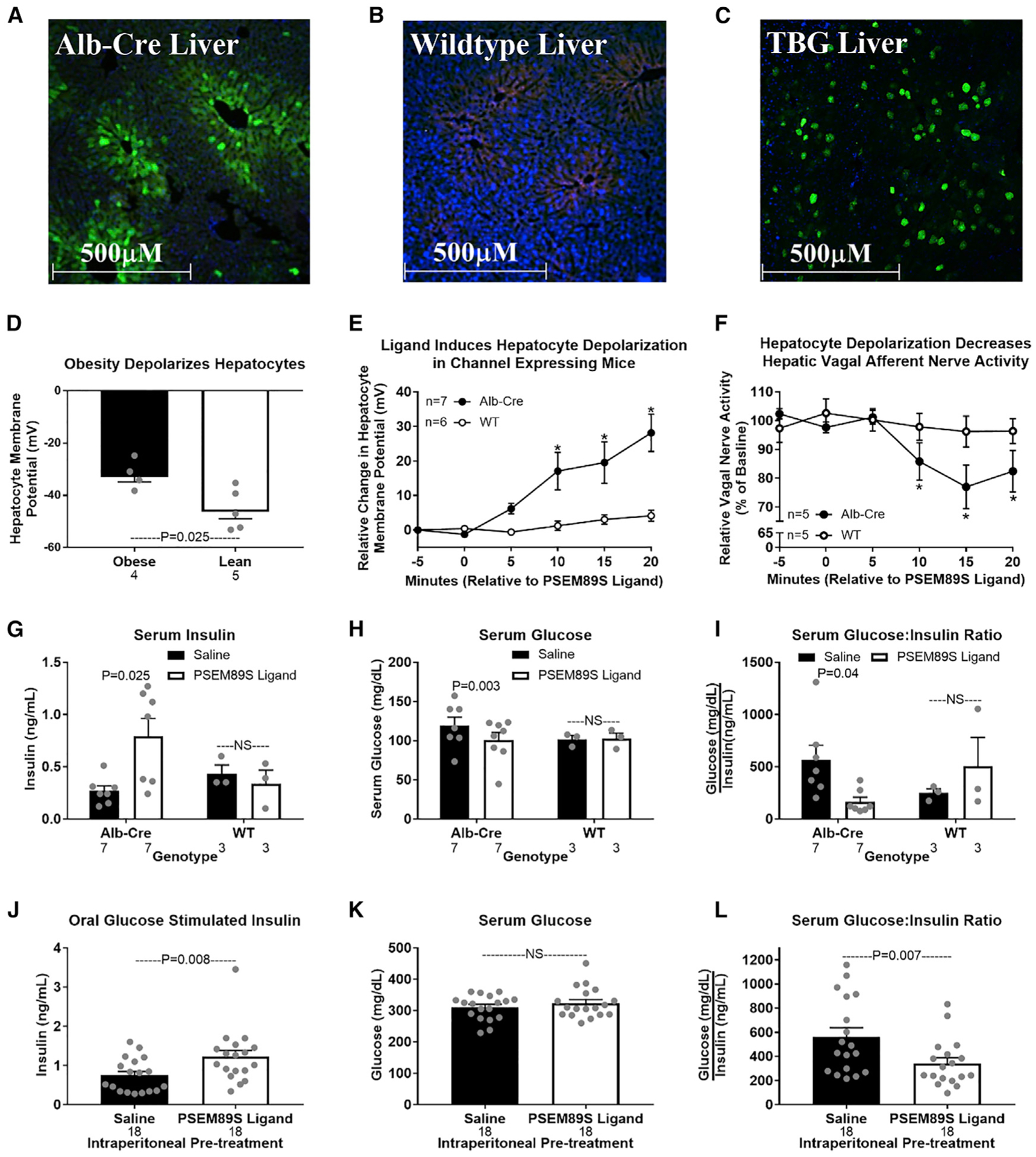
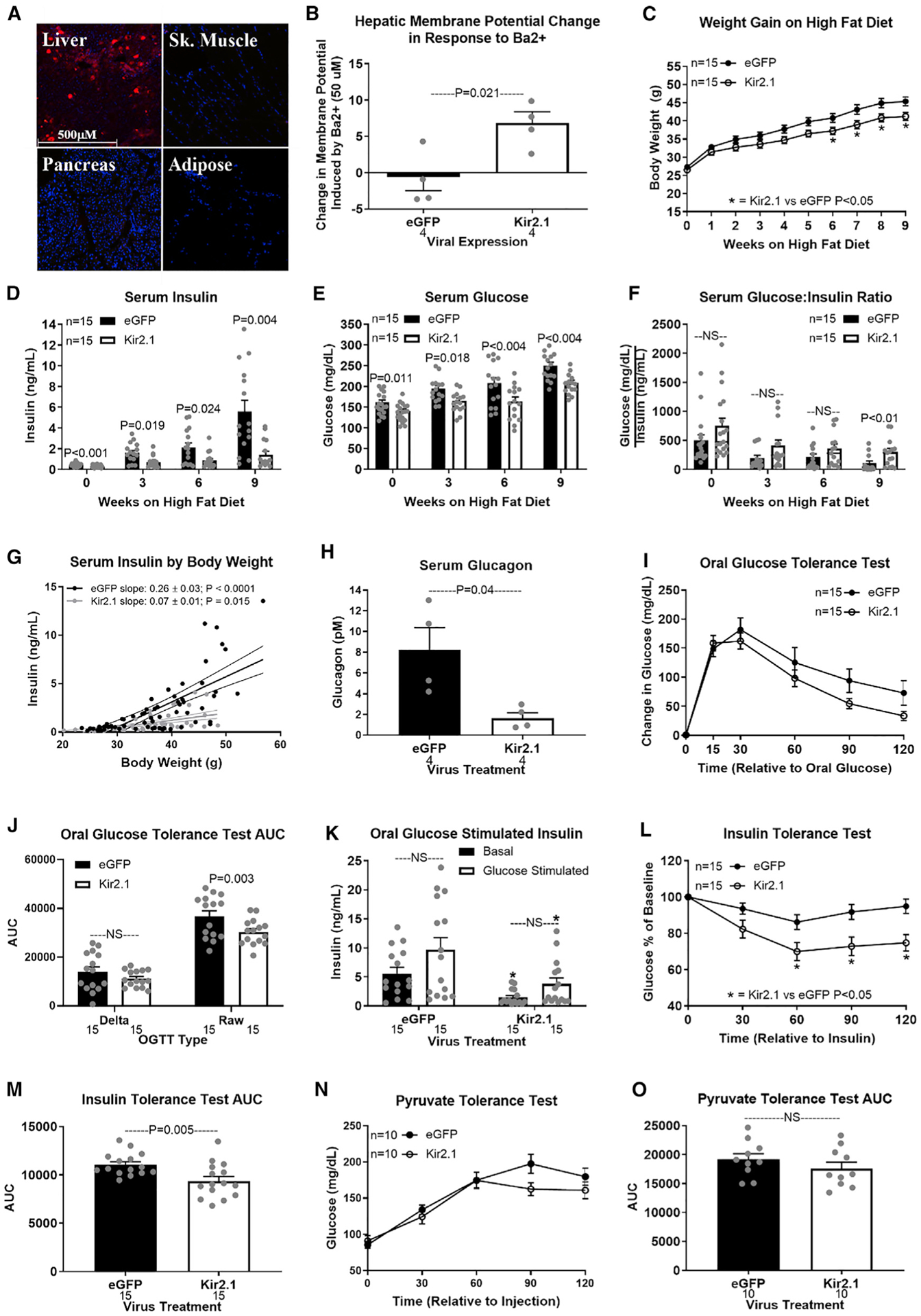
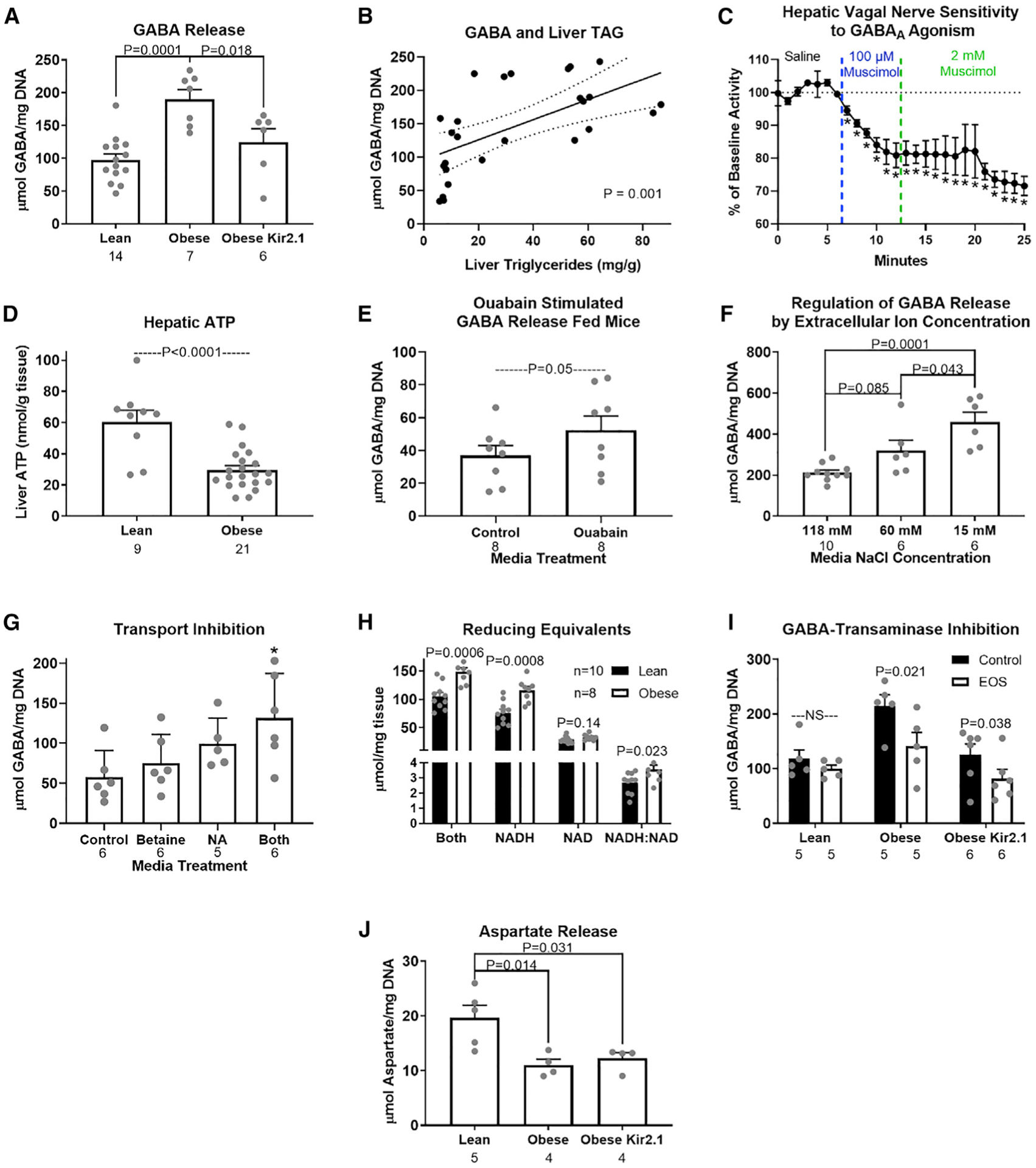
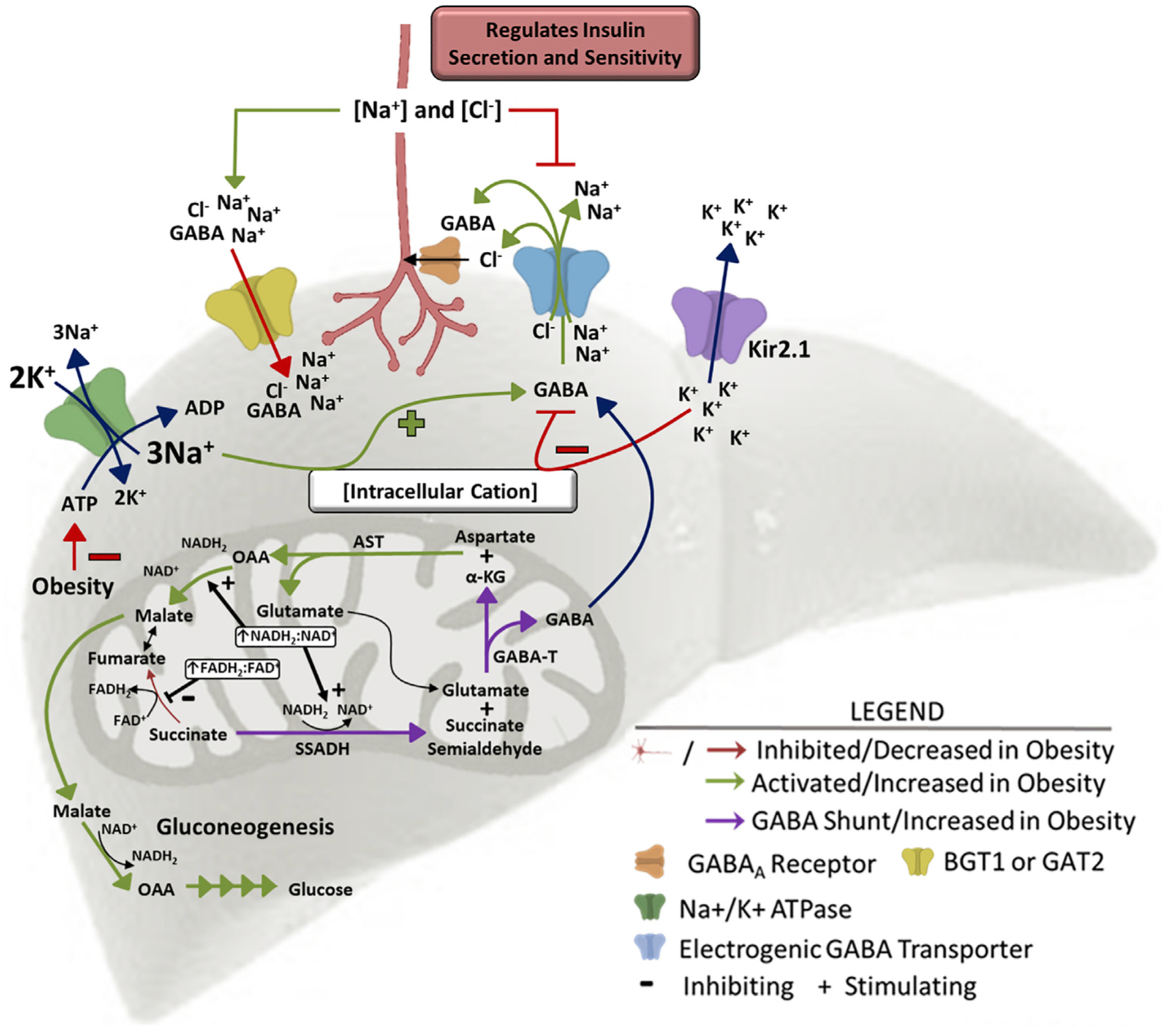
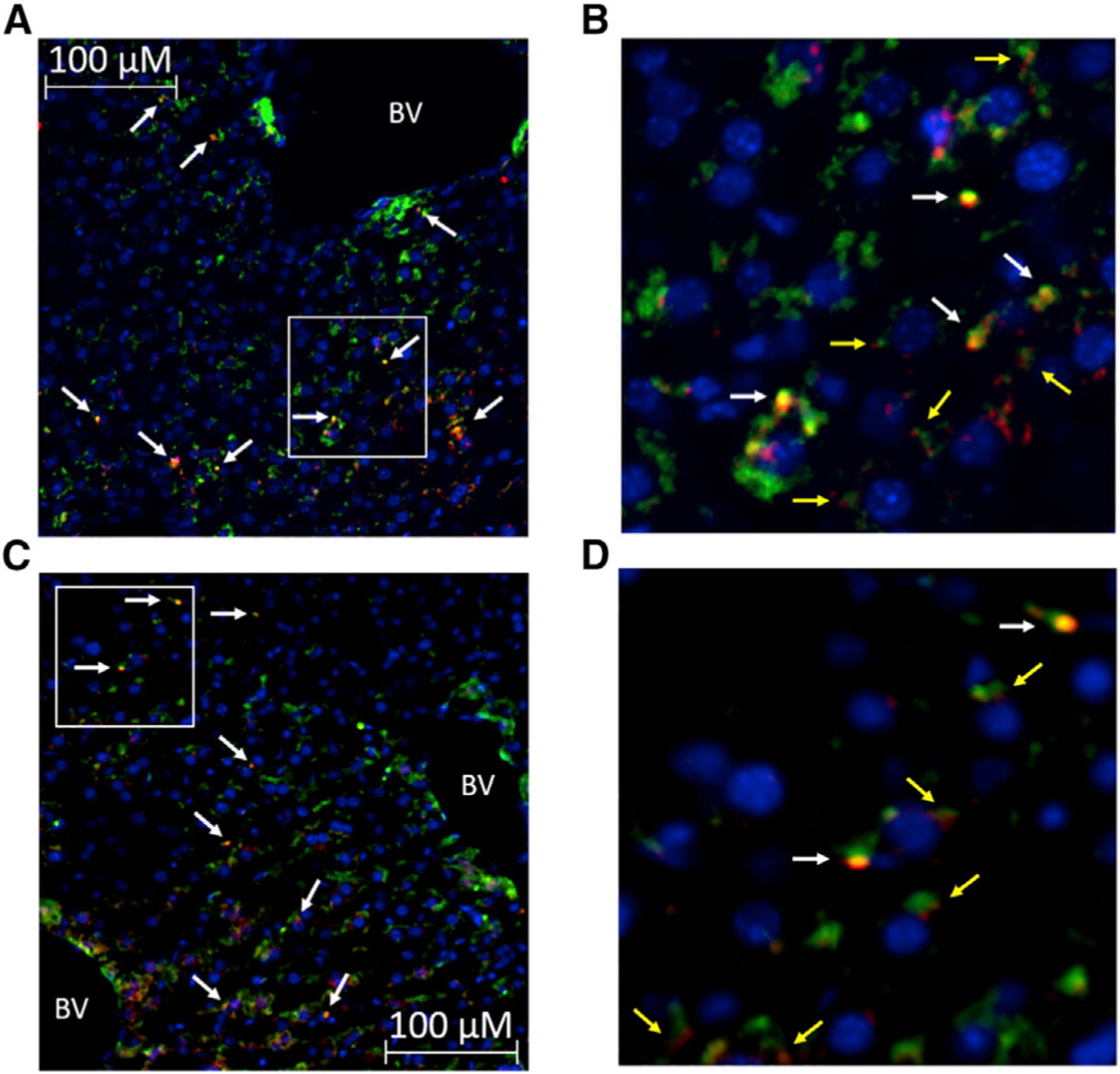
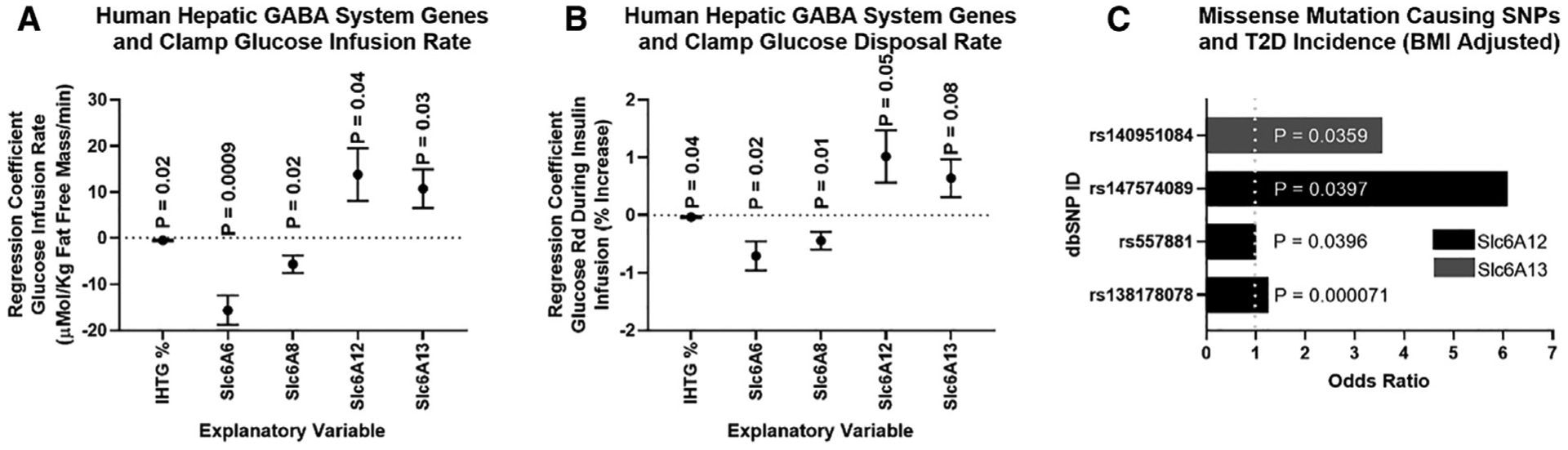
Hepatic lipid accumulation in obesity correlates with the severity of hyperinsulinemia and systemic insulin resistance. Obesity-induced hepatocellular lipid accumulation results in hepatocyte depolarization. We have established that hepatocyte depolarization depresses hepatic afferent vagal nerve firing, increases GABA release from liver slices, and causes hyperinsulinemia. Preventing hepatic GABA release or eliminating the ability of the liver to communicate to the hepatic vagal nerve ameliorates the hyperinsulinemia and insulin resistance associated with diet-induced obesity. In people with obesity, hepatic expression of GABA transporters is associated with glucose infusion and disposal rates during a hyperinsulinemic euglycemic clamp. Single-nucleotide polymorphisms in hepatic GABA re-uptake transporters are associated with an increased incidence of type 2 diabetes mellitus. Herein, we identify GABA as a neuro-hepatokine that is dysregulated in obesity and whose release can be manipulated to mute or exacerbate the glucoregulatory dysfunction common to obesity.
Keywords: GABA; GABA transporter; GABA-transaminase; NAFLD; Type 2 diabetes; hyperinsulinemia; insulin resistance; membrane potential; obesity.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The results presented in this paper have resulted in patent cooperation treaty Application No. 62/511,753 and 62/647,468: METHODS AND COMPOSITIONS FOR REGULATING GLUCOSE HOMEOSTASIS, which has been licensed by Livendocrine, LLC founded by Benjamin Renquist.
Figures
References
-
- Ahrén B, and Lundquist I (1982). Modulation of basal insulin secretion in the obese, hyperglycemic mouse. Metabolism 31, 172–179. - PubMed
-
- Balbo SL, Ribeiro RA, Mendes MC, Lubaczeuski C, Maller AC, Carneiro EM, and Bonfleur ML (2016). Vagotomy diminishes obesity in cafeteria rats by decreasing cholinergic potentiation of insulin release. J. Physiol. Biochem 72, 625–633. - PubMed
-
- Berthoud HR (2004). Anatomy and function of sensory hepatic nerves. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 280, 827–835. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
