

Integrative iTRAQ-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera
- PMID: 34193845
- PMCID: PMC8245520
- DOI: 10.1038/s41438-021-00591-2
Integrative iTRAQ-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera
Abstract
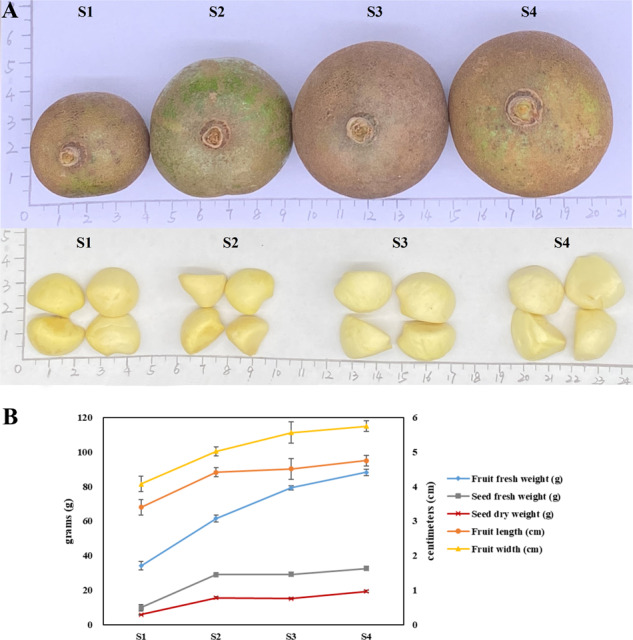
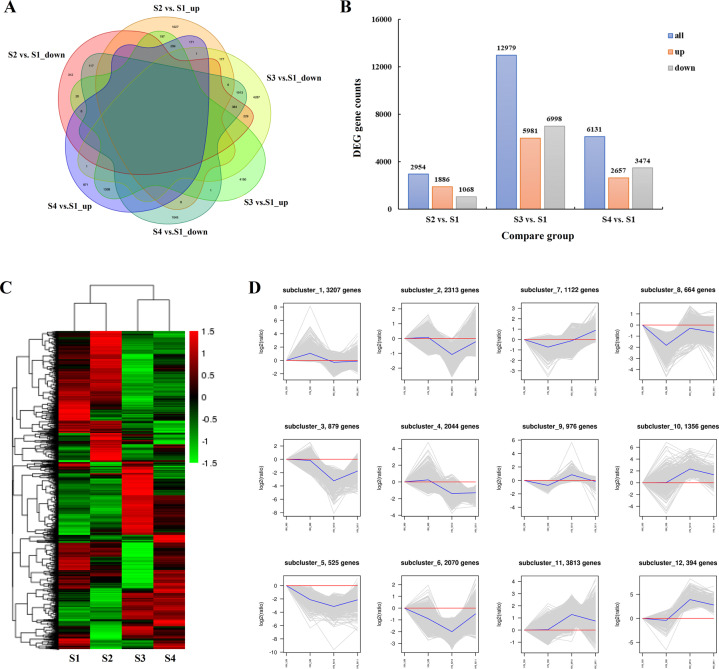
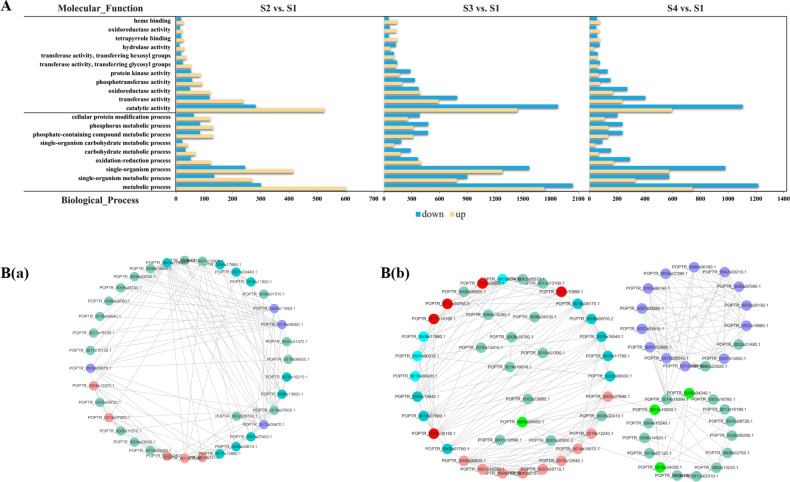
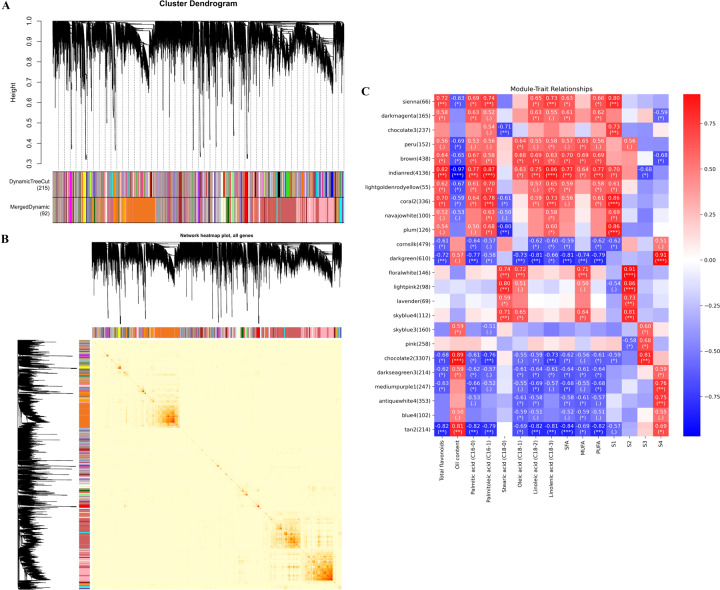
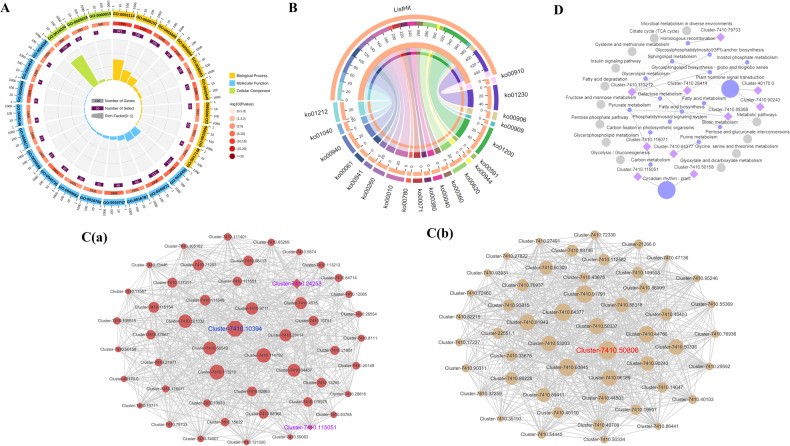
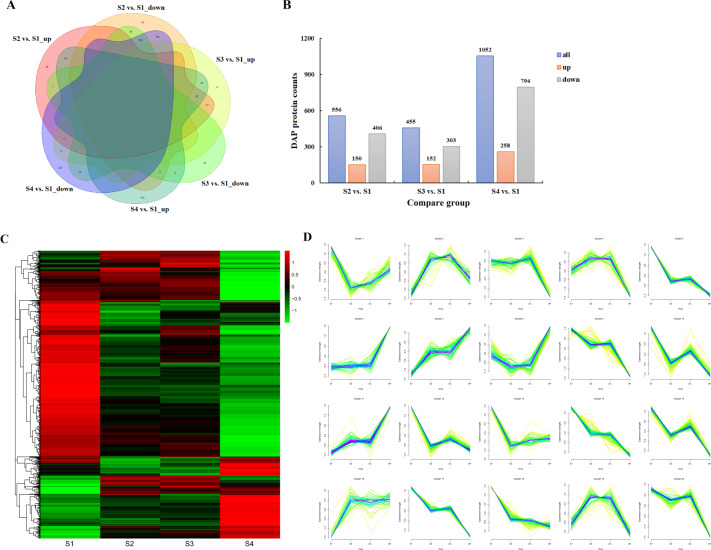
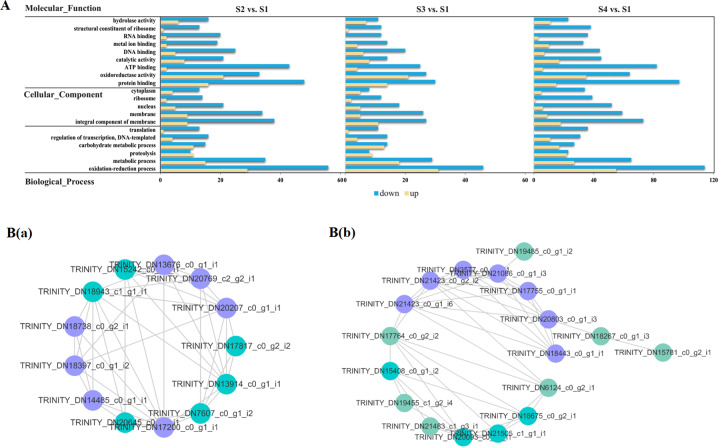
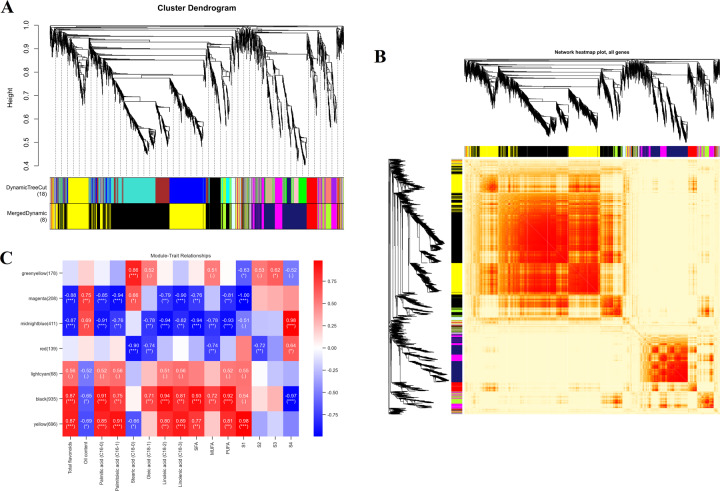
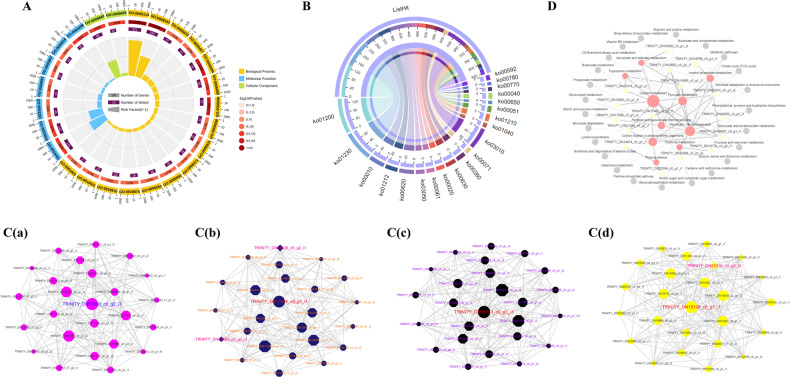
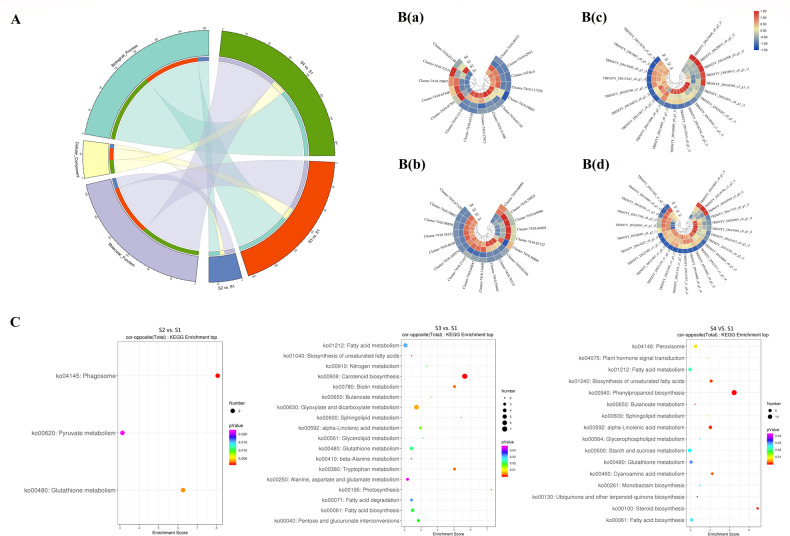
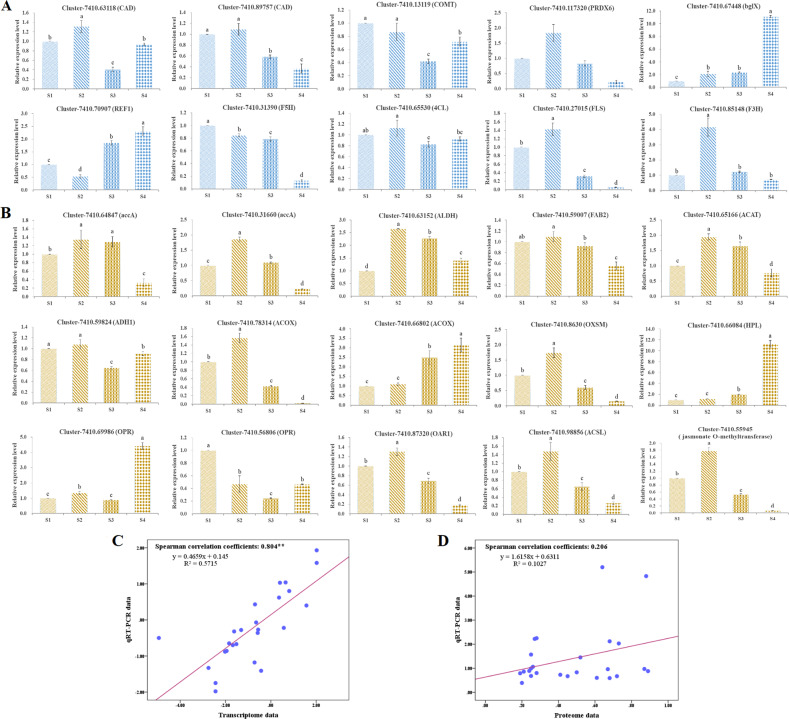
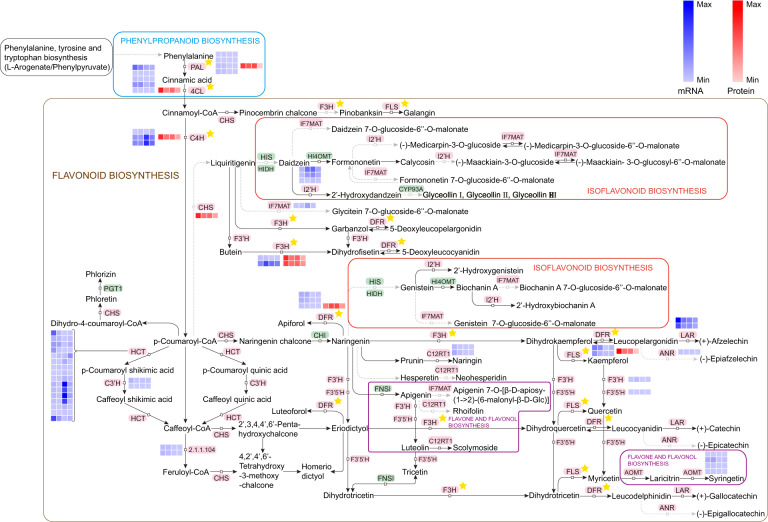
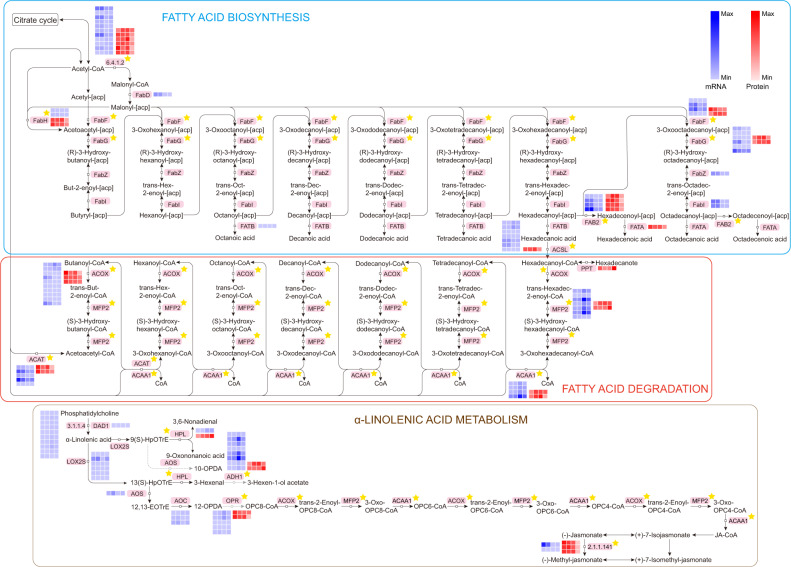
Camellia oleifera (C. oleifera) is one of the four major woody oil-bearing crops in the world and has relatively high ecological, economic, and medicinal value. Its seeds undergo a series of complex physiological and biochemical changes during ripening, which is mainly manifested as the accumulation and transformation of certain metabolites closely related to oil quality, especially flavonoids and fatty acids. To obtain new insights into the underlying molecular mechanisms, a parallel analysis of the transcriptome and proteome profiles of C. oleifera seeds at different maturity levels was conducted using RNA sequencing (RNA-seq) and isobaric tags for relative and absolute quantification (iTRAQ) complemented with gas chromatography-mass spectrometry (GC-MS) data. A total of 16,530 transcripts and 1228 proteins were recognized with significant differential abundances in pairwise comparisons of samples at various developmental stages. Among these, 317 were coexpressed with a poor correlation, and most were involved in metabolic processes, including fatty acid metabolism, α-linolenic acid metabolism, and glutathione metabolism. In addition, the content of total flavonoids decreased gradually with seed maturity, and the levels of fatty acids generally peaked at the fat accumulation stage; these results basically agreed with the regulation patterns of genes or proteins in the corresponding pathways. The expression levels of proteins annotated as upstream candidates of phenylalanine ammonia-lyase (PAL) and chalcone synthase (CHS) as well as their cognate transcripts were positively correlated with the variation in the flavonoid content, while shikimate O-hydroxycinnamoyltransferase (HCT)-encoding genes had the opposite pattern. The increase in the abundance of proteins and mRNAs corresponding to alcohol dehydrogenase (ADH) was associated with a reduction in linoleic acid synthesis. Using weighted gene coexpression network analysis (WGCNA), we further identified six unique modules related to flavonoid, oil, and fatty acid anabolism that contained hub genes or proteins similar to transcription factors (TFs), such as MADS intervening keratin-like and C-terminal (MIKC_MADS), type-B authentic response regulator (ARR-B), and basic helix-loop-helix (bHLH). Finally, based on the known metabolic pathways and WGCNA combined with the correlation analysis, five coexpressed transcripts and proteins composed of cinnamyl-alcohol dehydrogenases (CADs), caffeic acid 3-O-methyltransferase (COMT), flavonol synthase (FLS), and 4-coumarate: CoA ligase (4CL) were screened out. With this exploratory multiomics dataset, our results presented a dynamic picture regarding the maturation process of C. oleifera seeds on Hainan Island, not only revealing the temporal specific expression of key candidate genes and proteins but also providing a scientific basis for the genetic improvement of this tree species.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Jin XC. Bioactivities of water-soluble polysaccharides from fruit shell of Camellia oleifera Abel: antitumor and antioxidant activities. Carbohyd. Polym. 2012;87:2198–2201. doi: 10.1016/j.carbpol.2011.10.047. - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous
