

G-protein activation by a metabotropic glutamate receptor
- PMID: 34194039
- PMCID: PMC8822903
- DOI: 10.1038/s41586-021-03680-3
G-protein activation by a metabotropic glutamate receptor
Abstract
Family C G-protein-coupled receptors (GPCRs) operate as obligate dimers with extracellular domains that recognize small ligands, leading to G-protein activation on the transmembrane (TM) domains of these receptors by an unknown mechanism1. Here we show structures of homodimers of the family C metabotropic glutamate receptor 2 (mGlu2) in distinct functional states and in complex with heterotrimeric Gi. Upon activation of the extracellular domain, the two transmembrane domains undergo extensive rearrangement in relative orientation to establish an asymmetric TM6-TM6 interface that promotes conformational changes in the cytoplasmic domain of one protomer. Nucleotide-bound Gi can be observed pre-coupled to inactive mGlu2, but its transition to the nucleotide-free form seems to depend on establishing the active-state TM6-TM6 interface. In contrast to family A and B GPCRs, G-protein coupling does not involve the cytoplasmic opening of TM6 but is facilitated through the coordination of intracellular loops 2 and 3, as well as a critical contribution from the C terminus of the receptor. The findings highlight the synergy of global and local conformational transitions to facilitate a new mode of G-protein activation.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
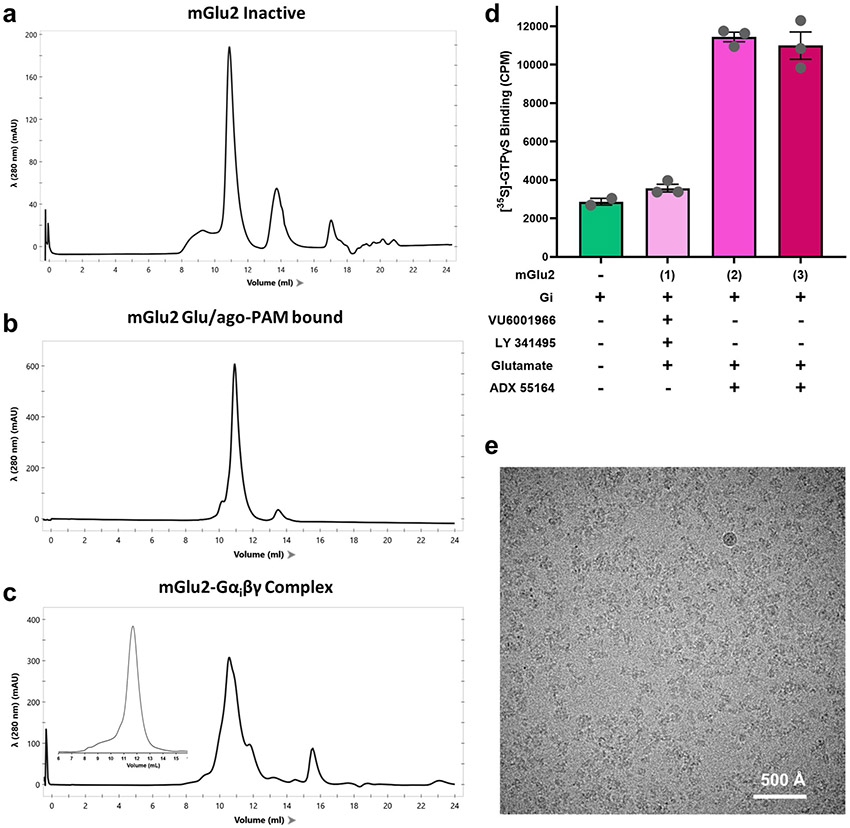




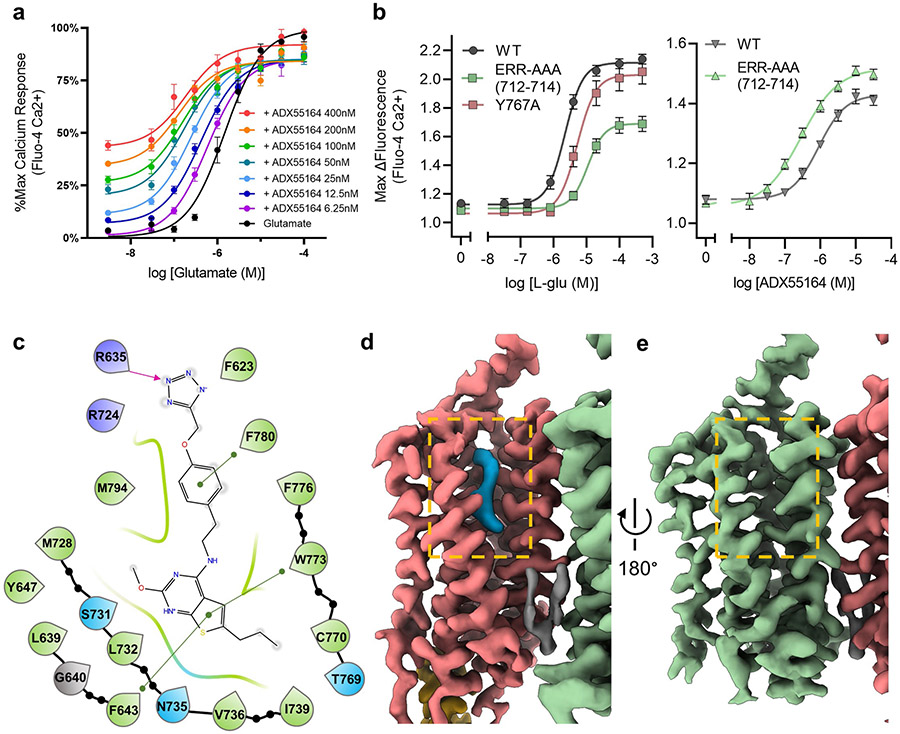






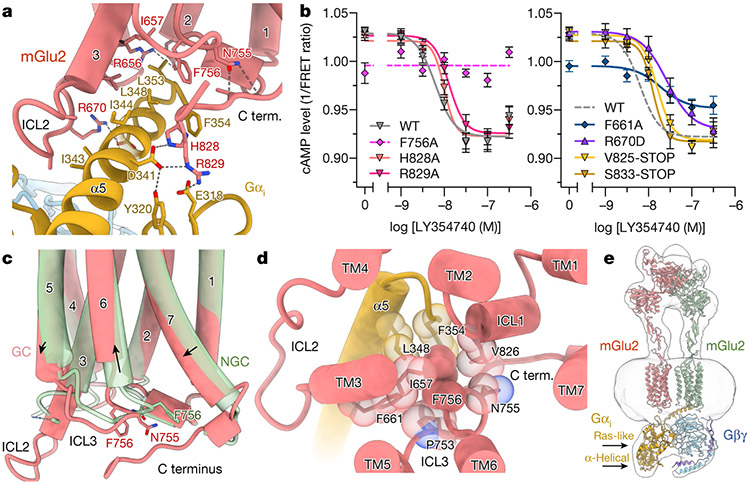

References
-
- Attwood TK & Findlay JB Fingerprinting G-protein-coupled receptors. Protein Eng. 7, 195–203 (1994). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
