

Hemophilic arthropathy: Current knowledge and future perspectives
- PMID: 34197690
- PMCID: PMC8456897
- DOI: 10.1111/jth.15444
Hemophilic arthropathy: Current knowledge and future perspectives
Abstract
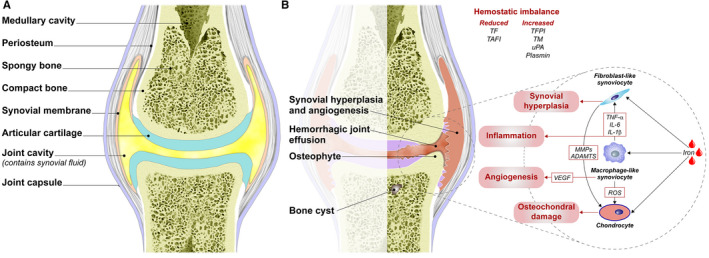
Hemophilia A and B are rare X-linked inherited bleeding disorders caused by complete or partial deficiency in or the absence of coagulation factors VIII and IX. Recurrent joint bleeding (hemarthrosis) is the most frequent clinical manifestation of severe hemophilia. Unless appropriately managed, even subclinical hemarthrosis can lead to the development of hemophilic arthropathy, a disabling condition characterized by joint remodelling, chronic pain, and a reduced quality of life, and eventually requires joint replacement. Given the lack of specific treatments to reduce blood-induced synovitis, the prevention of bleeding is pivotal to the maintenance of joint health. Prophylactic coagulation factor replacement therapy using extended half-life recombinant drugs has significantly improved patients' quality of life by reducing the burden of intravenous injections, and the more recent introduction of nonreplacement therapies such as subcutaneous emicizumab injections has improved treatment adherence and led to the greater protection of patients with hemophilia A. However, despite these advances, chronic arthropathy is still a significant problem. The introduction of point-of-care ultrasound imaging has improved the diagnosis of acute hemarthrosis and early hemophilic arthropathy, and allowed the better monitoring of progressive joint damage, but further research into the underlying mechanisms of the disease is required to allow the development of more targeted treatment. In the meantime, patient management should be based on the risk factors for the onset and progression of arthropathy of each individual patient, and all patients should be collaboratively cared for by multidisciplinary teams of hematologists, rheumatologists, orthopedic surgeons, and physiotherapists at comprehensive hemophilia treatment centers.
Keywords: hemarthrosis; hemophilia; hemophilic arthropathy; sub-clinical joint bleeding.
© 2021 International Society on Thrombosis and Haemostasis.
Conflict of interest statement
Dr. Gualtierotti is a member of advisory boards of Biomarin, Pfizer, Bayer, and Takeda, and participates in educational seminars sponsored by Pfizer, Sobi, and Roche. Dr. Peyvandi is a member of advisory boards of Bioverativ, Grifols, Roche, Sanofi, Sobi, Spark, and Takeda. Dr. Solimeno has no disclosure to make.
Figures
References
-
- Bolton‐Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801‐1809. - PubMed
-
- Di Minno MN, Ambrosino P, Franchini M, Coppola A, Di Minno G. Arthropathy in patients with moderate hemophilia a: a systematic review of the literature. Semin Thromb Hemost. 2013;39:723‐731. - PubMed
-
- Peyvandi F, Garagiola I, Biguzzi E. Advances in the treatment of bleeding disorders. J Thromb Haemost. 2016;14:2095‐2106. - PubMed
-
- Collins PW. Personalized prophylaxis. Haemophilia. 2012;18(Suppl 4):131‐135. - PubMed
