

BET Protein-Mediated Transcriptional Regulation in Heart Failure
- PMID: 34199719
- PMCID: PMC8199980
- DOI: 10.3390/ijms22116059
BET Protein-Mediated Transcriptional Regulation in Heart Failure
Abstract
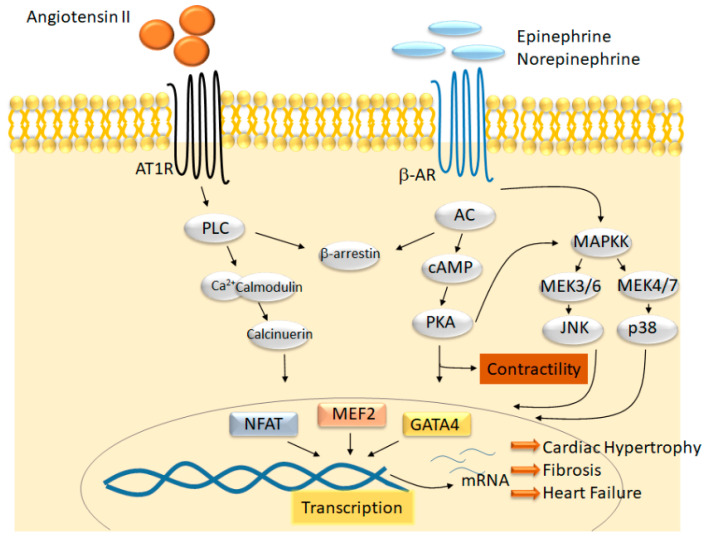
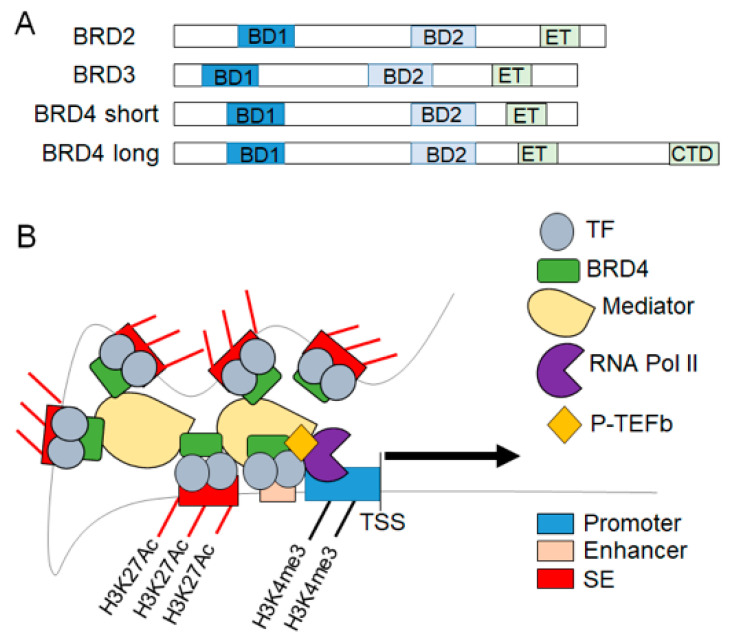
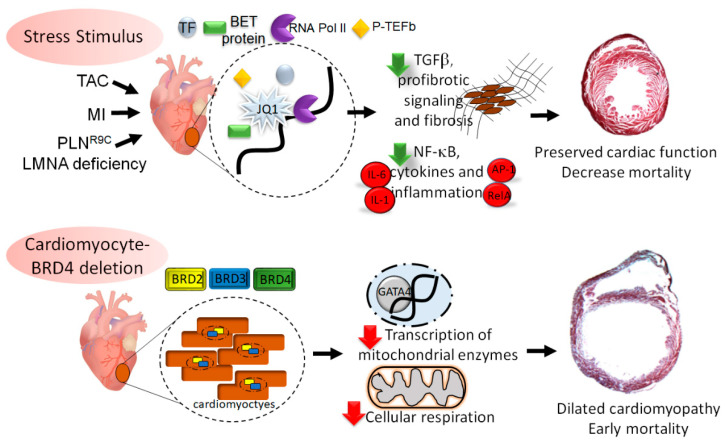
Heart failure is a complex disease process with underlying aberrations in neurohormonal systems that promote dysregulated cellular signaling and gene transcription. Over the past 10 years, the advent of small-molecule inhibitors that target transcriptional machinery has demonstrated the importance of the bromodomain and extraterminal (BET) family of epigenetic reader proteins in regulating gene transcription in multiple mouse models of cardiomyopathy. BETs bind to acetylated histone tails and transcription factors to integrate disparate stress signaling networks into a defined gene expression program. Under myocardial stress, BRD4, a BET family member, is recruited to superenhancers and promoter regions of inflammatory and profibrotic genes to promote transcription elongation. Whole-transcriptome analysis of BET-dependent gene networks suggests a major role of nuclear-factor kappa b and transforming growth factor-beta in the development of cardiac fibrosis and systolic dysfunction. Recent investigations also suggest a prominent role of BRD4 in maintaining cardiomyocyte mitochondrial respiration under basal conditions. In this review, we summarize the data from preclinical heart failure studies that explore the role of BET-regulated transcriptional mechanisms and delve into landmark studies that define BET bromodomain-independent processes involved in cardiac homeostasis.
Keywords: BET; BRD4; chromatin remodeling; dilated cardiomyopathy; heart failure; phospholamban; superenhancer; transcription regulation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
