

Usher Syndrome: Genetics of a Human Ciliopathy
- PMID: 34201633
- PMCID: PMC8268283
- DOI: 10.3390/ijms22136723
Usher Syndrome: Genetics of a Human Ciliopathy
Abstract
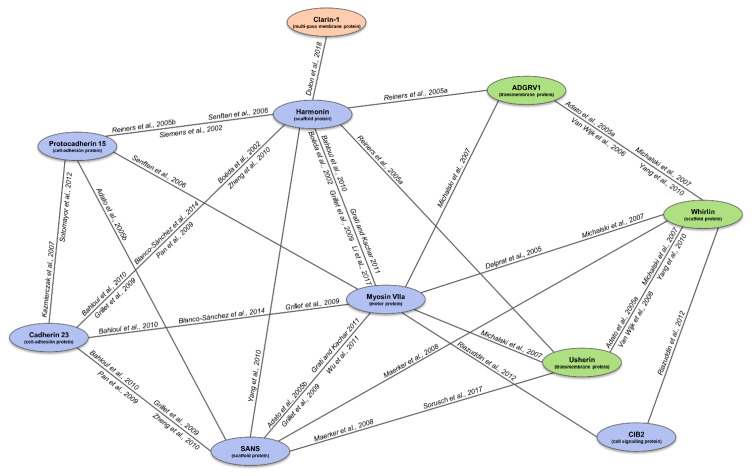
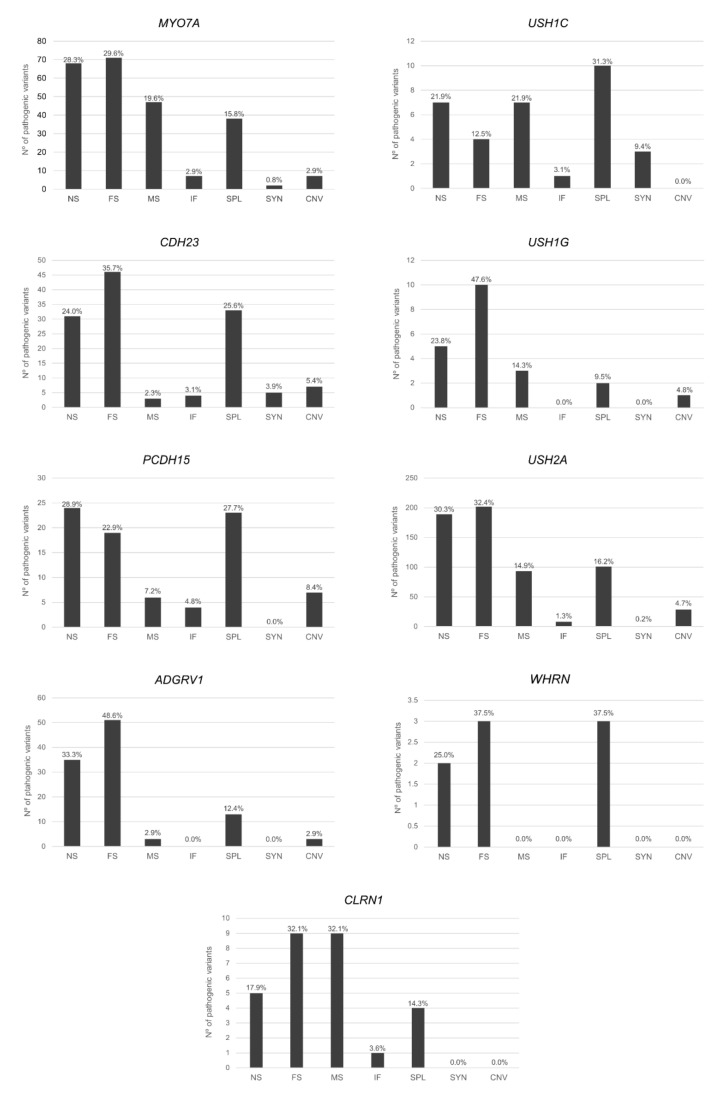
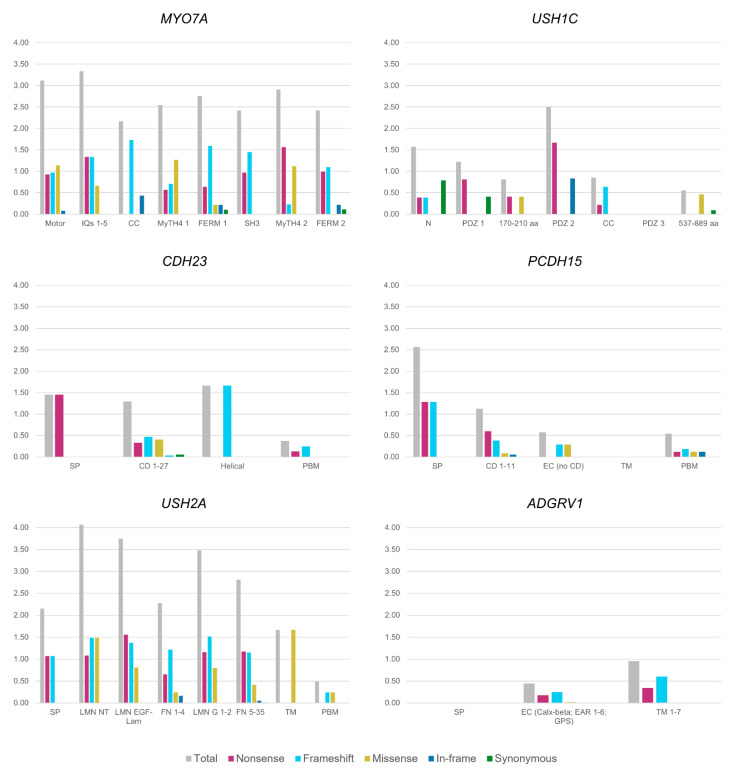
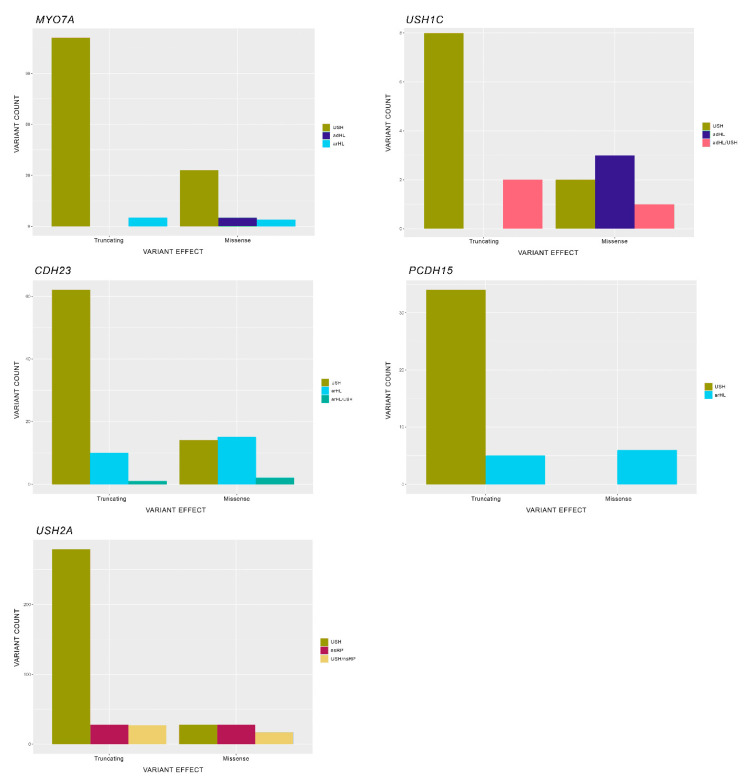
Usher syndrome (USH) is an autosomal recessive syndromic ciliopathy characterized by sensorineural hearing loss, retinitis pigmentosa and, sometimes, vestibular dysfunction. There are three clinical types depending on the severity and age of onset of the symptoms; in addition, ten genes are reported to be causative of USH, and six more related to the disease. These genes encode proteins of a diverse nature, which interact and form a dynamic protein network called the "Usher interactome". In the organ of Corti, the USH proteins are essential for the correct development and maintenance of the structure and cohesion of the stereocilia. In the retina, the USH protein network is principally located in the periciliary region of the photoreceptors, and plays an important role in the maintenance of the periciliary structure and the trafficking of molecules between the inner and the outer segments of photoreceptors. Even though some genes are clearly involved in the syndrome, others are controversial. Moreover, expression of some USH genes has been detected in other tissues, which could explain their involvement in additional mild comorbidities. In this paper, we review the genetics of Usher syndrome and the spectrum of mutations in USH genes. The aim is to identify possible mutation associations with the disease and provide an updated genotype-phenotype correlation.
Keywords: deafblindness; inherited retinal dystrophy; inner ear; pathogenic variant; photoreceptor; retinitis pigmentosa; sensorineural hearing loss; variant curation.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Davenport S.L.H., Omenn G.S. The Heterogeneity of Usher Syndrome; Proceedings of the 5th International Conference of Birth Defects; Montreal, QC, Canada. 21–27 August 1977.
-
- Bork J.M., Peters L., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R.S., et al. Usher Syndrome 1D and Nonsyndromic Autosomal Recessive Deafness DFNB12 Are Caused by Allelic Mutations of the Novel Cadherin-Like Gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
