

From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome
- PMID: 34201893
- PMCID: PMC8268977
- DOI: 10.3390/ijms22136767
From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome
Abstract
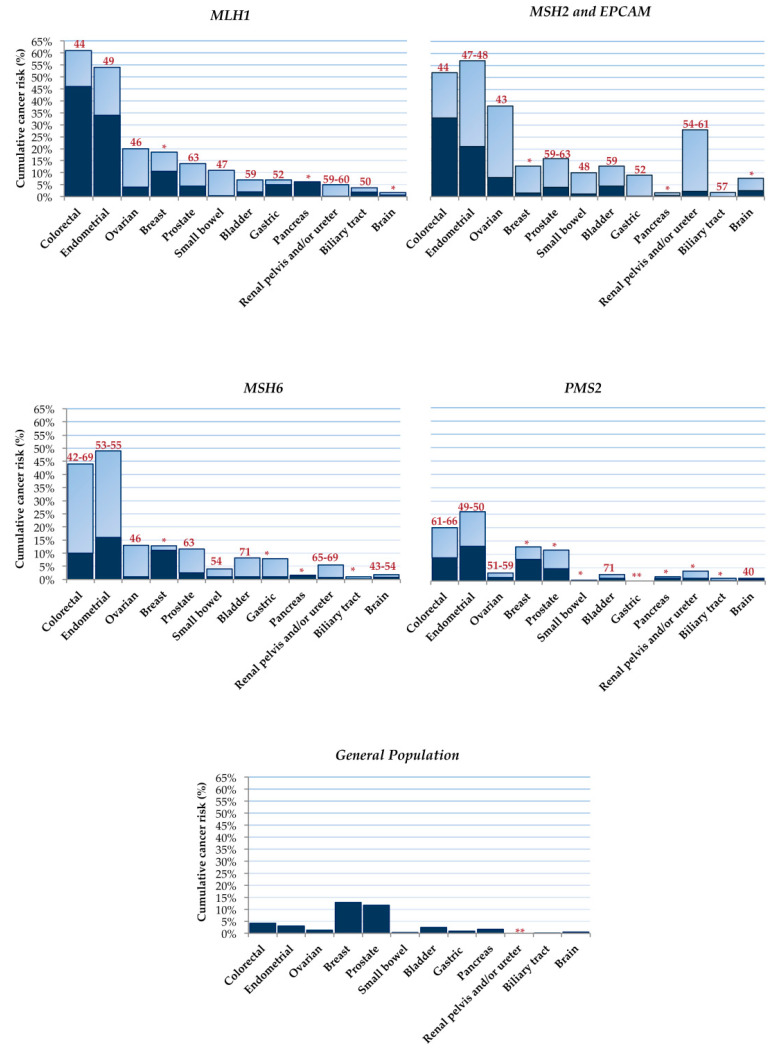
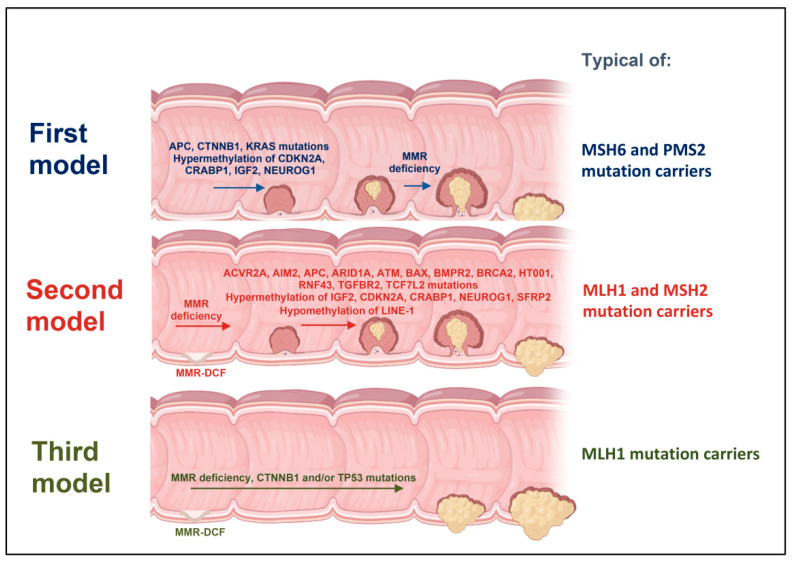
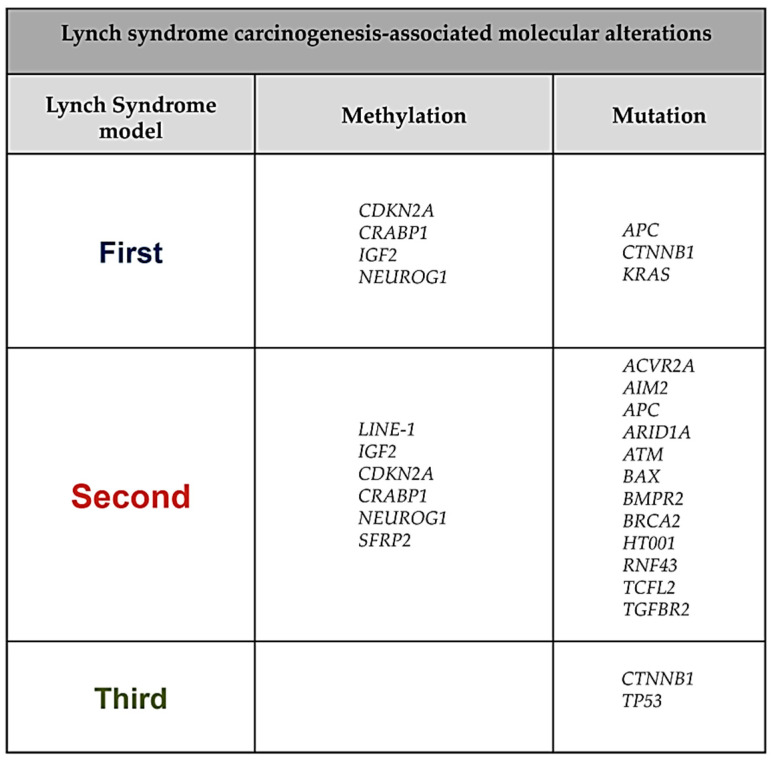
Lynch syndrome is a hereditary cancer-predisposing syndrome caused by germline defects in DNA mismatch repair (MMR) genes such as MLH1, MSH2, MSH6, and PMS2. Carriers of pathogenic mutations in these genes have an increased lifetime risk of developing colorectal cancer (CRC) and other malignancies. Despite intensive surveillance, Lynch patients typically develop CRC after 10 years of follow-up, regardless of the screening interval. Recently, three different molecular models of colorectal carcinogenesis were identified in Lynch patients based on when MMR deficiency is acquired. In the first pathway, adenoma formation occurs in an MMR-proficient background, and carcinogenesis is characterized by APC and/or KRAS mutation and IGF2, NEUROG1, CDK2A, and/or CRABP1 hypermethylation. In the second pathway, deficiency in the MMR pathway is an early event arising in macroscopically normal gut surface before adenoma formation. In the third pathway, which is associated with mutations in CTNNB1 and/or TP53, the adenoma step is skipped, with fast and invasive tumor growth occurring in an MMR-deficient context. Here, we describe the association between molecular and histological features in these three routes of colorectal carcinogenesis in Lynch patients. The findings summarized in this review may guide the use of individualized surveillance guidelines based on a patient's carcinogenesis subtype.
Keywords: CRC; Lynch syndrome; MMR genes; early detection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Stoffel E.M., Turgeon D.K., Stockwell D.H., Zhao L., Normolle D.P., Tuck M.K., Bresalier R.S., Marcon N.E., Baron J.A., Ruffin M.T., et al. Missed Adenomas during Colonoscopic Surveillance in Individuals with Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) Cancer Prev. Res. 2008;1:470–475. doi: 10.1158/1940-6207.CAPR-08-0098. - DOI - PMC - PubMed
-
- Liu T., Yan H., Kuismanen S., Percesepe A., Bisgaard M.L., Pedroni M., Benatti P., Kinzler K.W., Vogelstein B., Ponz de Leon M., et al. The Role of HPMS1 and HPMS2 in Predisposing to Colorectal Cancer. Cancer Res. 2001;61:7798–7802. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
