

Cystic Fibrosis Human Organs-on-a-Chip
- PMID: 34202364
- PMCID: PMC8305167
- DOI: 10.3390/mi12070747
Cystic Fibrosis Human Organs-on-a-Chip
Abstract
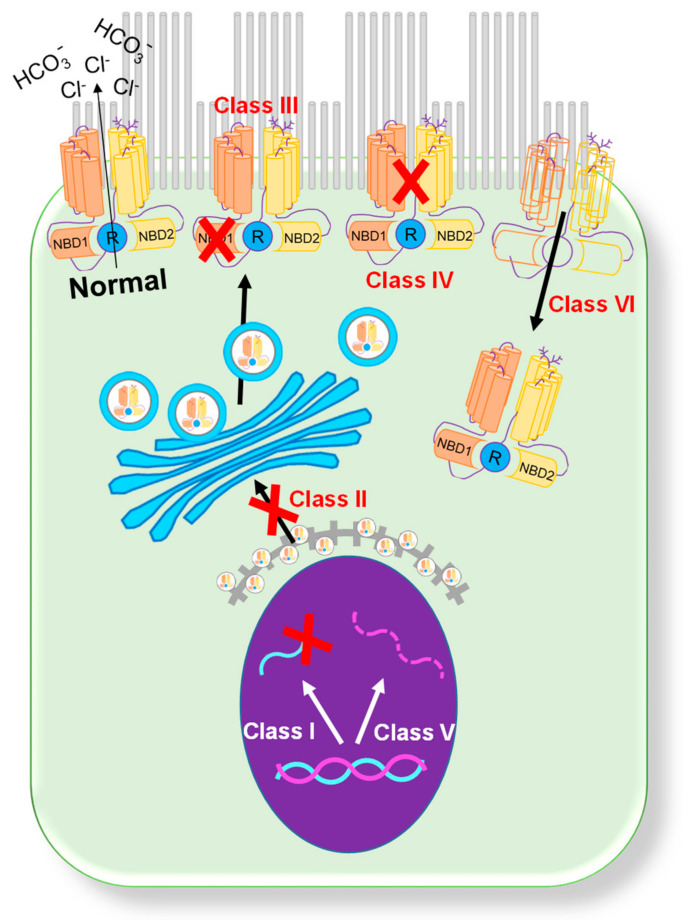
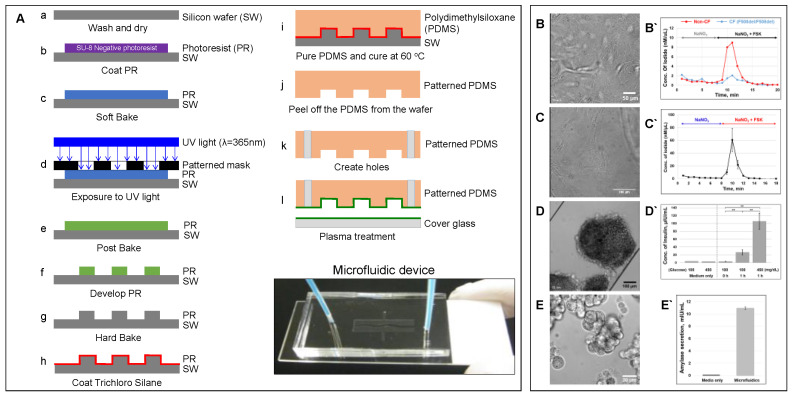
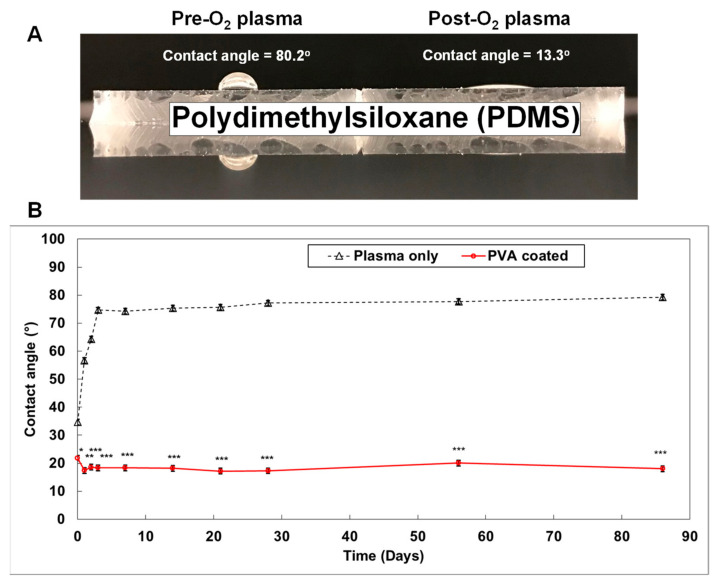
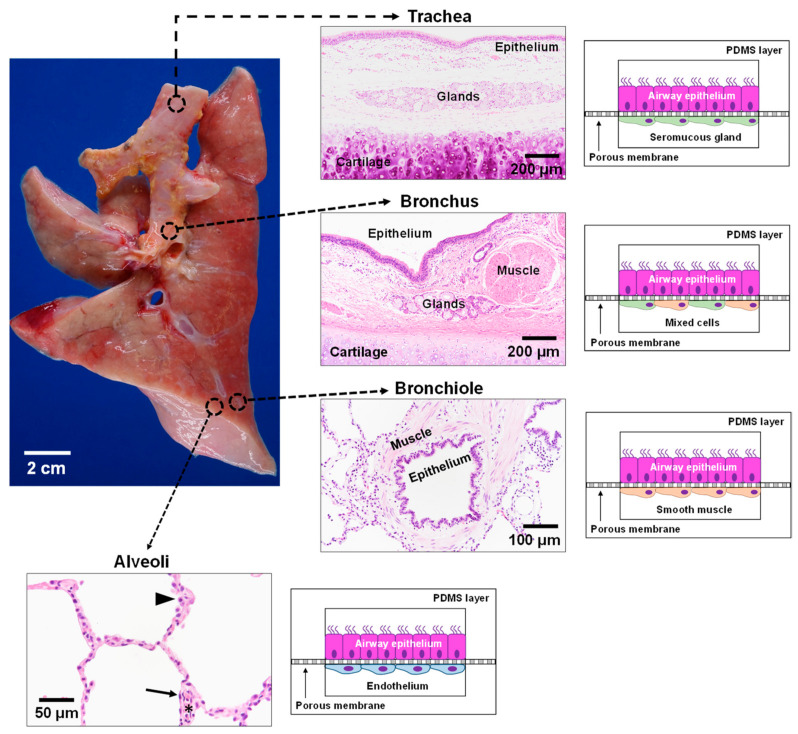
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene: the gene product responsible for transporting chloride and bicarbonate ions through the apical membrane of most epithelial cells. Major clinical features of CF include respiratory failure, pancreatic exocrine insufficiency, and intestinal disease. Many CF animal models have been generated, but some models fail to fully capture the phenotypic manifestations of human CF disease. Other models that better capture the key characteristics of the human CF phenotype are cost prohibitive or require special care to maintain. Important differences have been reported between the pathophysiology seen in human CF patients and in animal models. These limitations present significant limitations to translational research. This review outlines the study of CF using patient-derived organs-on-a-chip to overcome some of these limitations. Recently developed microfluidic-based organs-on-a-chip provide a human experimental model that allows researchers to manipulate environmental factors and mimic in vivo conditions. These chips may be scaled to support pharmaceutical studies and may also be used to study organ systems and human disease. The use of these chips in CF discovery science enables researchers to avoid the barriers inherent in animal models and promote the advancement of personalized medicine.
Keywords: CFTR; cystic fibrosis; organ-on-a-chip; personalized medicine.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
[Relation between gene mutations and pancreatic exocrine function in patients with cystic fibrosis].Srp Arh Celok Lek. 2001 May-Jun;129 Suppl 1:6-9. Srp Arh Celok Lek. 2001. PMID: 15637983 Serbian.
-
Genotype and phenotype in cystic fibrosis.Respiration. 2000;67(2):117-33. doi: 10.1159/000029497. Respiration. 2000. PMID: 10773783 Review.
-
Personalized medicine in CF: from modulator development to therapy for cystic fibrosis patients with rare CFTR mutations.Am J Physiol Lung Cell Mol Physiol. 2018 Apr 1;314(4):L529-L543. doi: 10.1152/ajplung.00465.2017. Epub 2017 Dec 14. Am J Physiol Lung Cell Mol Physiol. 2018. PMID: 29351449 Free PMC article. Review.
-
State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy.Pharmaceuticals (Basel). 2021 Sep 15;14(9):928. doi: 10.3390/ph14090928. Pharmaceuticals (Basel). 2021. PMID: 34577628 Free PMC article. Review.
-
Expression of delta F508 cystic fibrosis transmembrane conductance regulator protein and related chloride transport properties in the gallbladder epithelium from cystic fibrosis patients.Hepatology. 1999 Jun;29(6):1624-34. doi: 10.1002/hep.510290634. Hepatology. 1999. PMID: 10347100
Cited by
-
Gene Polymorphism of Biotransformation Enzymes and Ciprofloxacin Pharmacokinetics in Pediatric Patients with Cystic Fibrosis.Biomedicines. 2022 May 2;10(5):1050. doi: 10.3390/biomedicines10051050. Biomedicines. 2022. PMID: 35625789 Free PMC article.
-
Recent Developments in Inertial and Centrifugal Microfluidic Systems along with the Involved Forces for Cancer Cell Separation: A Review.Sensors (Basel). 2023 Jun 2;23(11):5300. doi: 10.3390/s23115300. Sensors (Basel). 2023. PMID: 37300027 Free PMC article. Review.
-
Human organoids-on-chips for biomedical research and applications.Theranostics. 2024 Jan 1;14(2):788-818. doi: 10.7150/thno.90492. eCollection 2024. Theranostics. 2024. PMID: 38169573 Free PMC article. Review.
-
Therapeutic Interventions for Pseudomonas Infections in Cystic Fibrosis Patients: A Review of Phase IV Trials.J Clin Med. 2024 Oct 30;13(21):6530. doi: 10.3390/jcm13216530. J Clin Med. 2024. PMID: 39518670 Free PMC article. Review.
-
Preclinical testing of antimicrobials for cystic fibrosis lung infections: current needs and future priorities.Microbiology (Reading). 2023 Jul;169(7):001361. doi: 10.1099/mic.0.001361. Microbiology (Reading). 2023. PMID: 37428539 Free PMC article.
References
-
- Andersen D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938;56:344–399. doi: 10.1001/archpedi.1938.01980140114013. - DOI
-
- Veit G., Avramescu R.G., Chiang A.N., Houck S.A., Cai Z., Peters K.W., Hong J.S., Pollard H.B., Guggino W.B., Balch W.E., et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016;27:424–433. doi: 10.1091/mbc.e14-04-0935. - DOI - PMC - PubMed
-
- Harutyunyan M., Huang Y.J., Mun K.S., Yang F.M.Y., Arora K., Naren A.P. Personalized medicine in CF: From modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018;314:1529–1543. doi: 10.1152/ajplung.00465.2017. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
