

From RNA World to SARS-CoV-2: The Edited Story of RNA Viral Evolution
- PMID: 34202997
- PMCID: PMC8234929
- DOI: 10.3390/cells10061557
From RNA World to SARS-CoV-2: The Edited Story of RNA Viral Evolution
Abstract
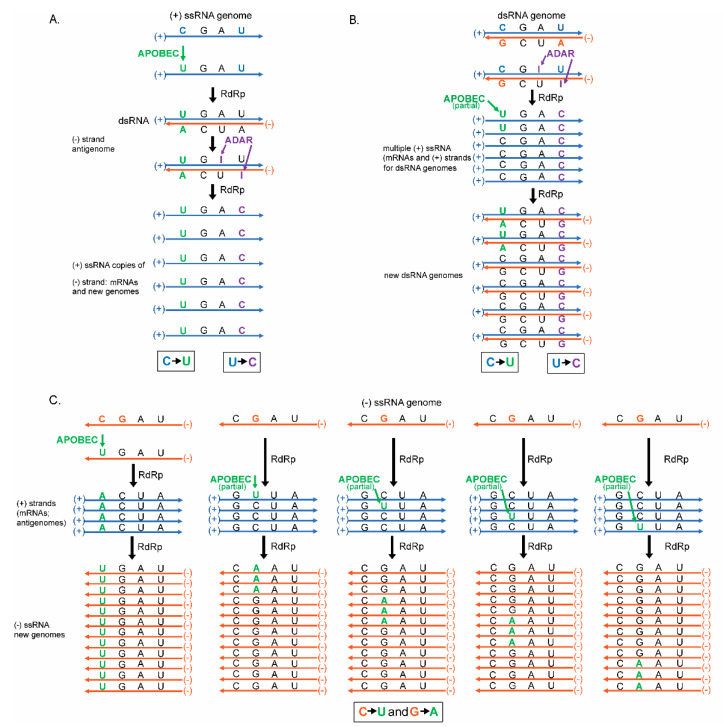
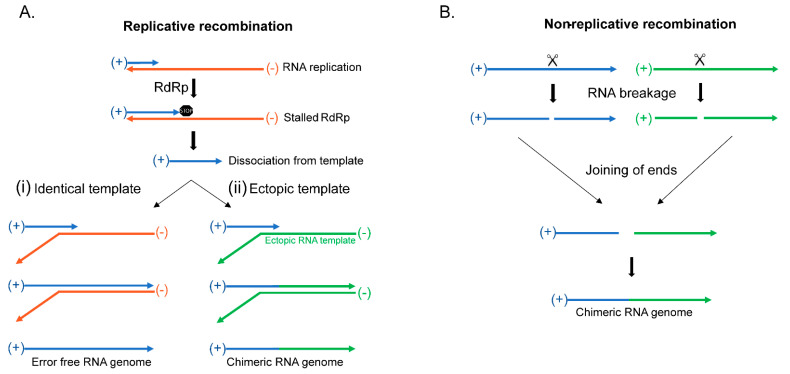
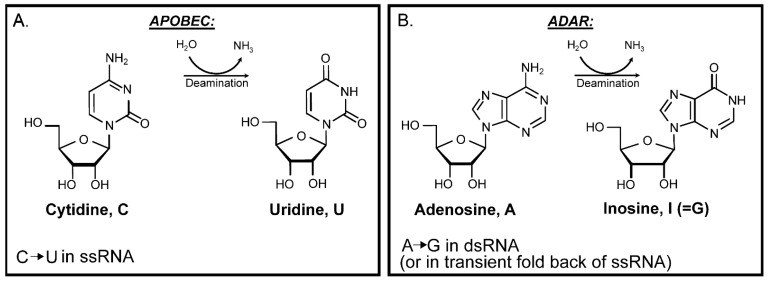
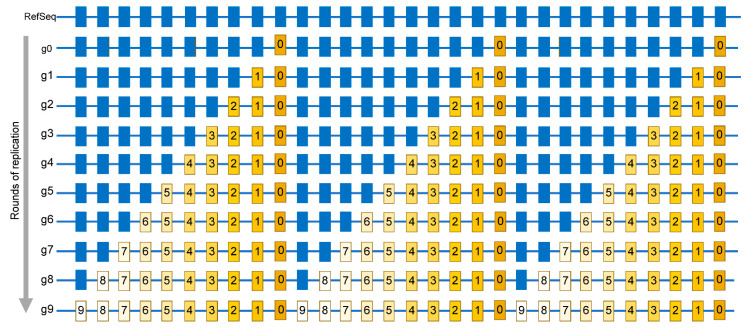
The current SARS-CoV-2 pandemic underscores the importance of understanding the evolution of RNA genomes. While RNA is subject to the formation of similar lesions as DNA, the evolutionary and physiological impacts RNA lesions have on viral genomes are yet to be characterized. Lesions that may drive the evolution of RNA genomes can induce breaks that are repaired by recombination or can cause base substitution mutagenesis, also known as base editing. Over the past decade or so, base editing mutagenesis of DNA genomes has been subject to many studies, revealing that exposure of ssDNA is subject to hypermutation that is involved in the etiology of cancer. However, base editing of RNA genomes has not been studied to the same extent. Recently hypermutation of single-stranded RNA viral genomes have also been documented though its role in evolution and population dynamics. Here, we will summarize the current knowledge of key mechanisms and causes of RNA genome instability covering areas from the RNA world theory to the SARS-CoV-2 pandemic of today. We will also highlight the key questions that remain as it pertains to RNA genome instability, mutations accumulation, and experimental strategies for addressing these questions.
Keywords: ADAR; APOBEC; RNA editing; RNA world theory; genome stability; hypermutation; messenger RNA; viral RNA; viral evolution.
Conflict of interest statement
Both listed Authors have declared no competing interests.
Figures

References
-
- Dobzhansky T. Nothing in Biology Makes Sense except in the Light of Evolution. Am. Biol. Teach. 1973;35:125–129. doi: 10.2307/4444260. - DOI
-
- Dobzhansky T. Genetics and the Origin of Species/Theodosius Dobzhansky. 3rd ed. Columbia University Press; New York, NY, USA: 1951.
-
- Domingo E. Virus as Populations. Academic Press; Boston, MA, USA: 2016. Molecular Basis of Genetic Variation of Viruses; pp. 35–71. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
