

Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer
- PMID: 34204306
- PMCID: PMC8234537
- DOI: 10.3390/biom11060901
Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer
Abstract
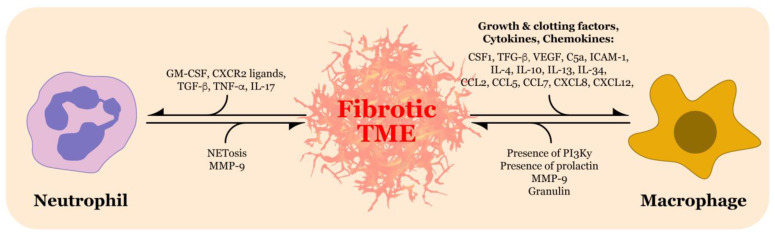
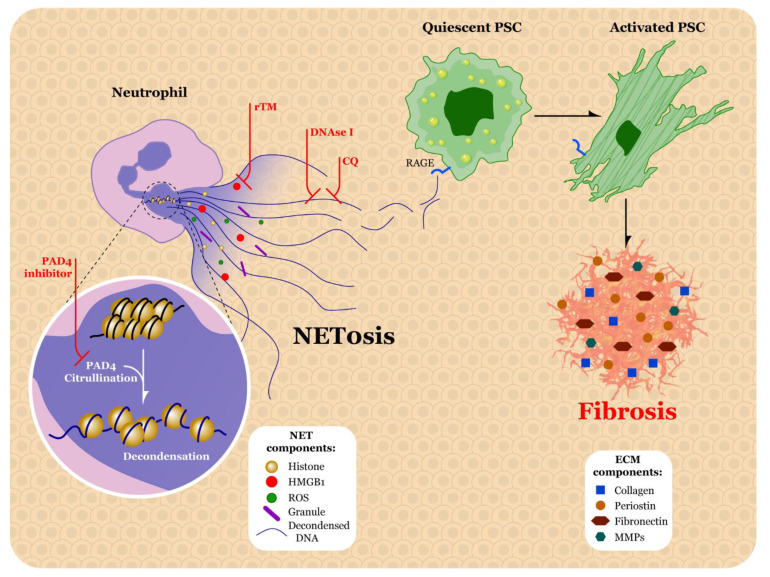
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy with a five-year survival rate of only 9%. PDAC is characterized by a dense, fibrotic stroma composed of extracellular matrix (ECM) proteins. This desmoplastic stroma is a hallmark of PDAC, representing a significant physical barrier that is immunosuppressive and obstructs penetration of cytotoxic chemotherapy agents into the tumor microenvironment (TME). Additionally, dense ECM promotes hypoxia, making tumor cells refractive to radiation therapy and alters their metabolism, thereby supporting proliferation and survival. In this review, we outline the significant contribution of fibrosis to the pathogenesis of pancreatic cancer, with a focus on the cross talk between immune cells and pancreatic stellate cells that contribute to ECM deposition. We emphasize the cellular mechanisms by which neutrophils and macrophages, specifically, modulate the ECM in favor of PDAC-progression. Furthermore, we investigate how activated stellate cells and ECM influence immune cells and promote immunosuppression in PDAC. Finally, we summarize therapeutic strategies that target the stroma and hinder immune cell promotion of fibrogenesis, which have unfortunately led to mixed results. An enhanced understanding of the complex interactions between the pancreatic tumor ECM and immune cells may uncover novel treatment strategies that are desperately needed for this devastating disease.
Keywords: extracellular matrix; fibrosis; immune cell modulation; macrophages; neutrophil extracellular trap; neutrophils; pancreatic ductal adenocarcinoma.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Pancreatic stellate cells and the interleukin family: Linking fibrosis and immunity to pancreatic ductal adenocarcinoma (Review).Mol Med Rep. 2024 Sep;30(3):159. doi: 10.3892/mmr.2024.13283. Epub 2024 Jul 12. Mol Med Rep. 2024. PMID: 38994764 Free PMC article. Review.
-
Pancreatic Tumor Microenvironment.Adv Exp Med Biol. 2020;1296:243-257. doi: 10.1007/978-3-030-59038-3_15. Adv Exp Med Biol. 2020. PMID: 34185297
-
Fibrotic Fortresses and Therapeutic Frontiers: Pancreatic Stellate Cells and the Extracellular Matrix in Pancreatic Cancer.Cancer Med. 2025 Jun;14(11):e70788. doi: 10.1002/cam4.70788. Cancer Med. 2025. PMID: 40437741 Free PMC article. Review.
-
Pancreatic Stellate Cells Promote Tumor Progression by Promoting an Immunosuppressive Microenvironment in Murine Models of Pancreatic Cancer.Pancreas. 2020 Jan;49(1):120-127. doi: 10.1097/MPA.0000000000001464. Pancreas. 2020. PMID: 31856087
-
Sono-promoted drug penetration and extracellular matrix modulation potentiate sonodynamic therapy of pancreatic ductal adenocarcinoma.Acta Biomater. 2023 Apr 15;161:265-274. doi: 10.1016/j.actbio.2023.02.038. Epub 2023 Mar 7. Acta Biomater. 2023. PMID: 36893956
Cited by
-
Metabolic Pathways as a Novel Landscape in Pancreatic Ductal Adenocarcinoma.Cancers (Basel). 2022 Aug 4;14(15):3799. doi: 10.3390/cancers14153799. Cancers (Basel). 2022. PMID: 35954462 Free PMC article. Review.
-
Quantitative characterization of the 3D self-organization of PDAC tumor spheroids reveals cell type and matrix dependence through advanced microscopy analysis.APL Bioeng. 2025 Mar 27;9(1):016116. doi: 10.1063/5.0242490. eCollection 2025 Mar. APL Bioeng. 2025. PMID: 40161492 Free PMC article.
-
Pancreatic stellate cells and the interleukin family: Linking fibrosis and immunity to pancreatic ductal adenocarcinoma (Review).Mol Med Rep. 2024 Sep;30(3):159. doi: 10.3892/mmr.2024.13283. Epub 2024 Jul 12. Mol Med Rep. 2024. PMID: 38994764 Free PMC article. Review.
-
Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer.Cancers (Basel). 2021 Dec 31;14(1):194. doi: 10.3390/cancers14010194. Cancers (Basel). 2021. PMID: 35008355 Free PMC article. Review.
-
Cancer stem cell-derived extracellular vesicles preferentially target MHC-II-macrophages and PD1+ T cells in the tumor microenvironment.PLoS One. 2023 Feb 3;18(2):e0279400. doi: 10.1371/journal.pone.0279400. eCollection 2023. PLoS One. 2023. PMID: 36735677 Free PMC article.
References
-
- Tavakkoli A., Singal A.G., Waljee A.K., Elmunzer B.J., Pruitt S.L., McKey T., Rubenstein J.H., Scheiman J.M., Murphy C.C. Racial disparities and trends in pancreatic cancer incidence and mortality in the united states. Clin. Gastroenterol. Hepatol. 2020;18:171–178.e110. doi: 10.1016/j.cgh.2019.05.059. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
