

Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives
- PMID: 34204594
- PMCID: PMC8231124
- DOI: 10.3390/antiox10060950
Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives
Abstract
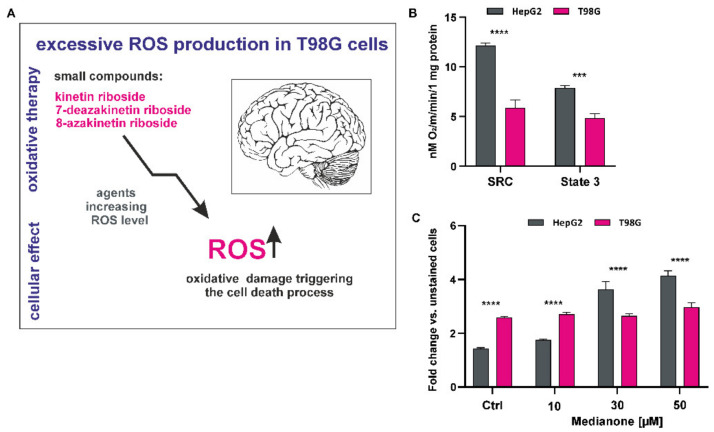
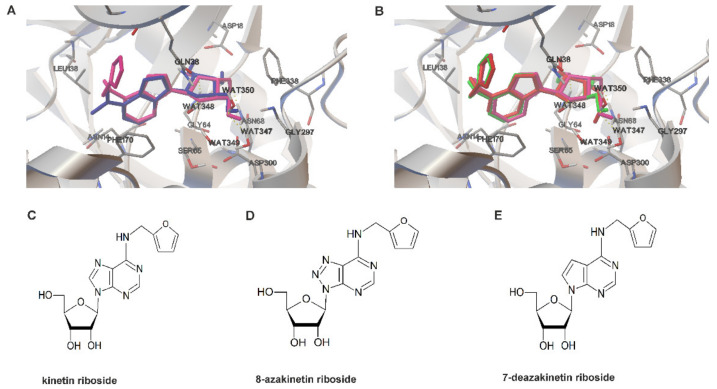
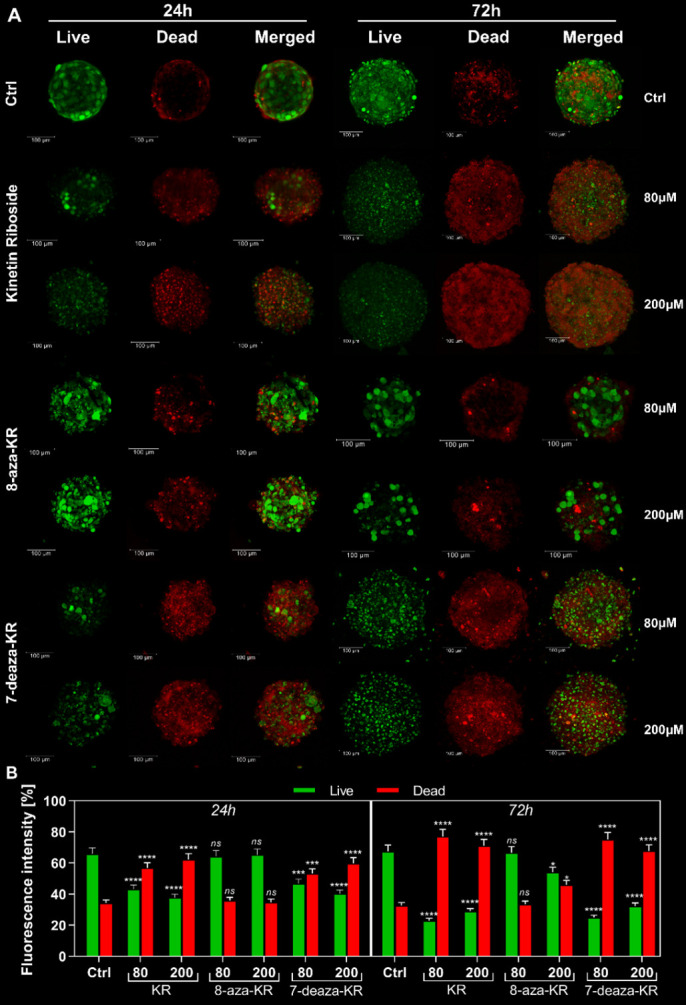
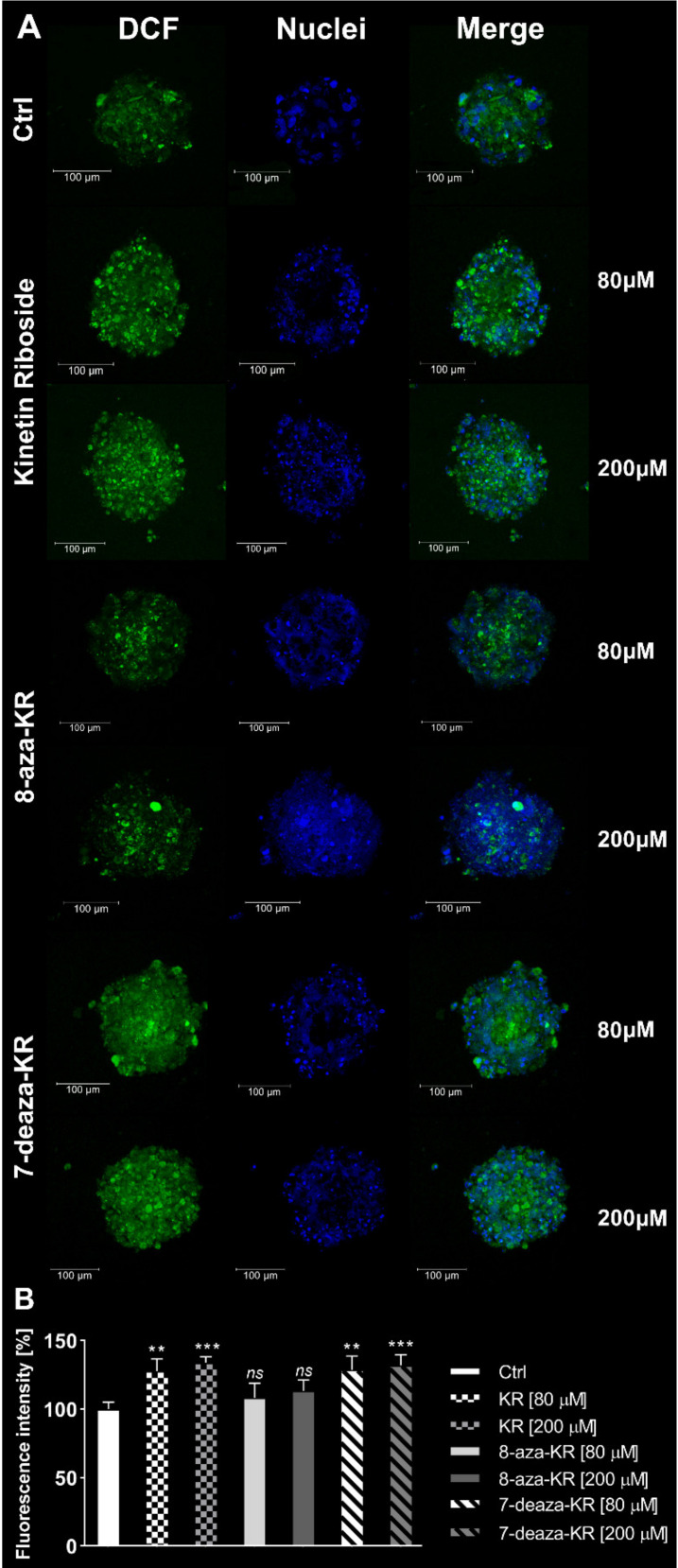
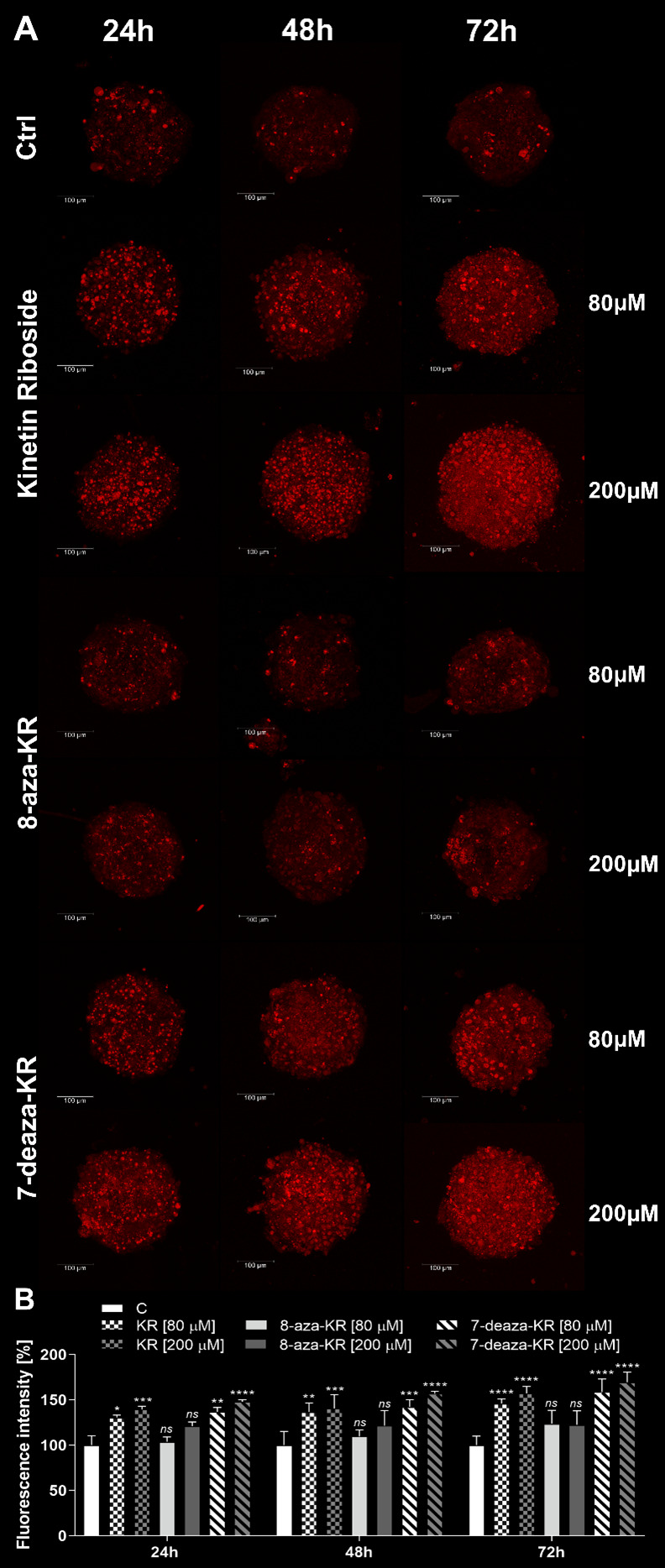
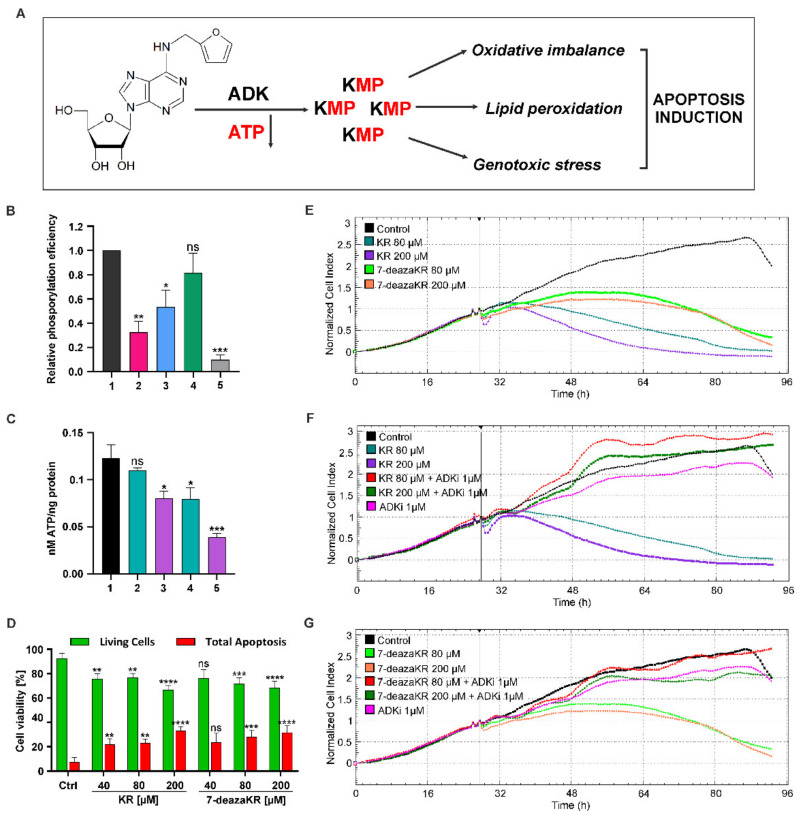
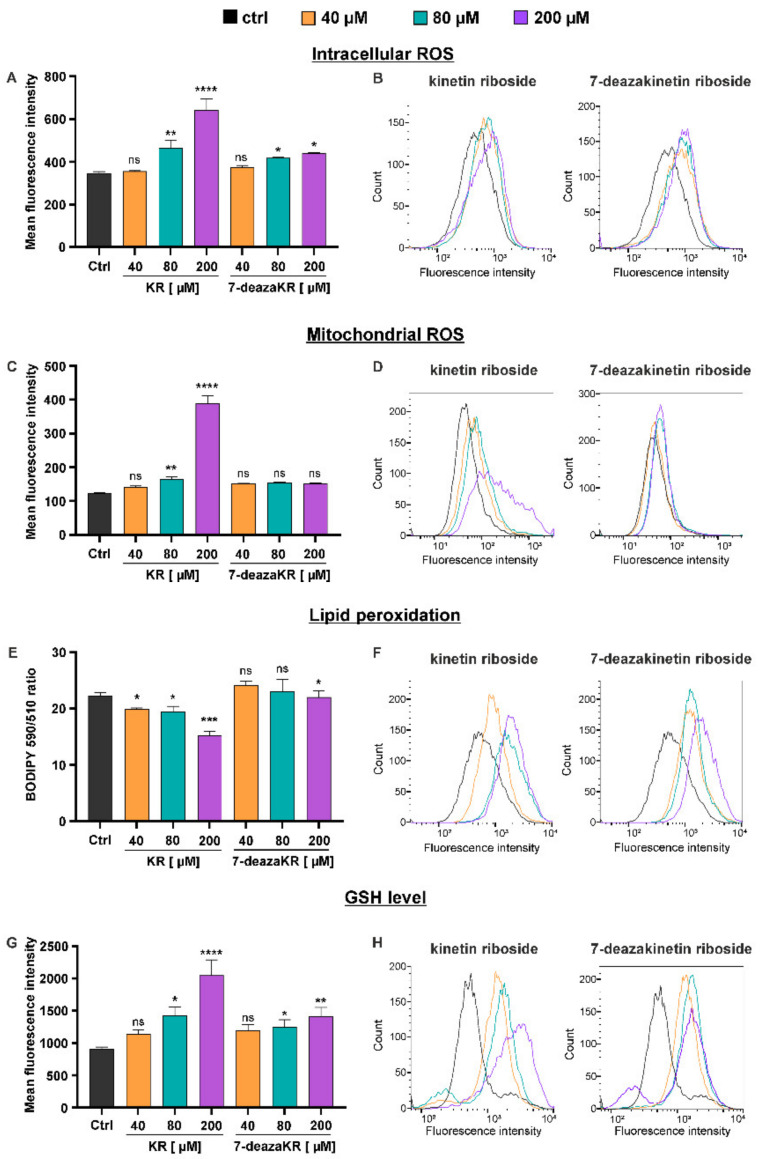
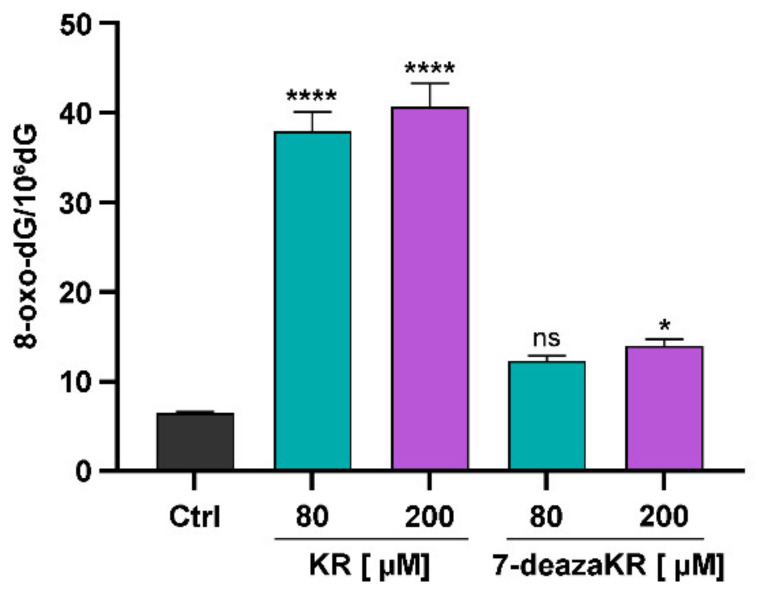
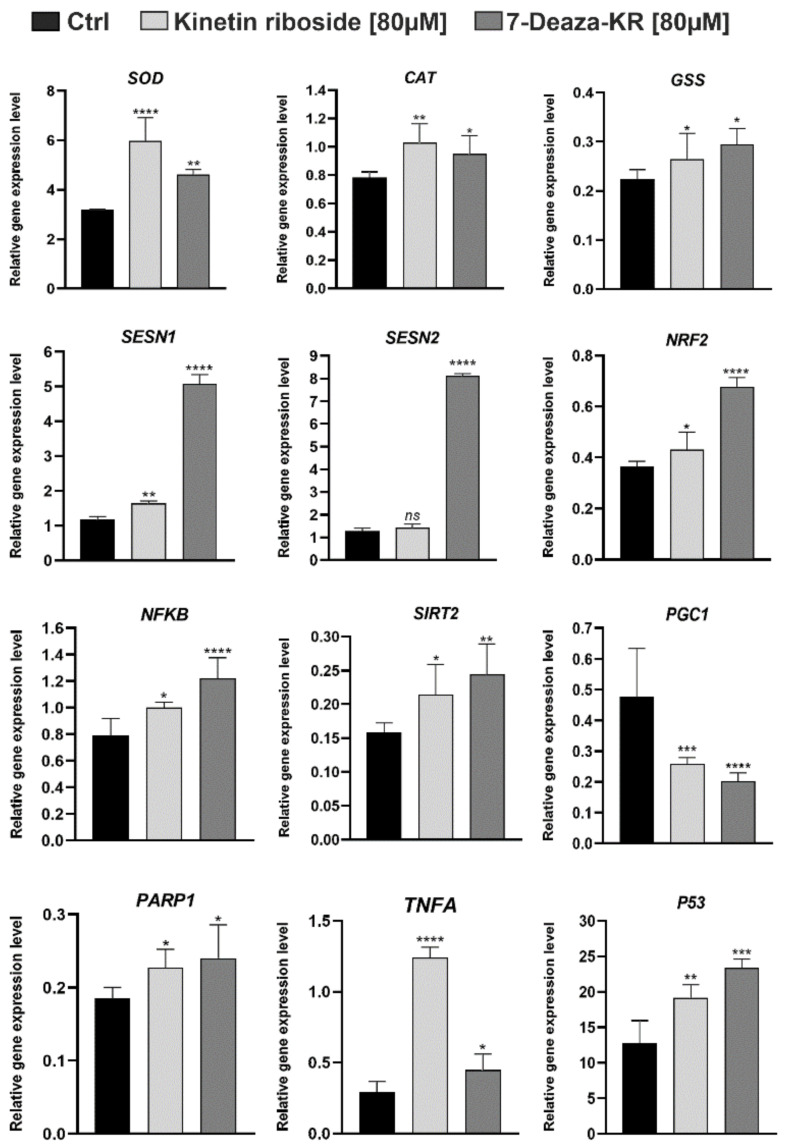
Recently, small compound-based therapies have provided new insights into the treatment of glioblastoma multiforme (GBM) by inducing oxidative impairment. Kinetin riboside (KR) and newly designed derivatives (8-azaKR, 7-deazaKR) selectively affect the molecular pathways crucial for cell growth by interfering with the redox status of cancer cells. Thus, these compounds might serve as potential alternatives in the oxidative therapy of GBM. The increased basal levels of reactive oxygen species (ROS) in GBM support the survival of cancer cells and cause drug resistance. The simplest approach to induce cell death is to achieve the redox threshold and circumvent the antioxidant defense mechanisms. Consequently, cells become more sensitive to oxidative stress (OS) caused by exogenous agents. Here, we investigated the effect of KR and its derivatives on the redox status of T98G cells in 2D and 3D cell culture. The use of spheroids of T98G cells enabled the selection of one derivative-7-deazaKR-with comparable antitumor activity to KR. Both compounds induced ROS generation and genotoxic OS, resulting in lipid peroxidation and leading to apoptosis. Taken together, these results demonstrated that KR and 7-deazaKR modulate the cellular redox environment of T98G cells, and vulnerability of these cells is dependent on their antioxidant capacity.
Keywords: ROS; cancer cells; cell death; glioblastoma multiforme; kinetin riboside; oxidative imbalance; oxidative therapy; purine derivatives; spheroids.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
