

Altered Bone Status in Rett Syndrome
- PMID: 34205017
- PMCID: PMC8230033
- DOI: 10.3390/life11060521
Altered Bone Status in Rett Syndrome
Abstract
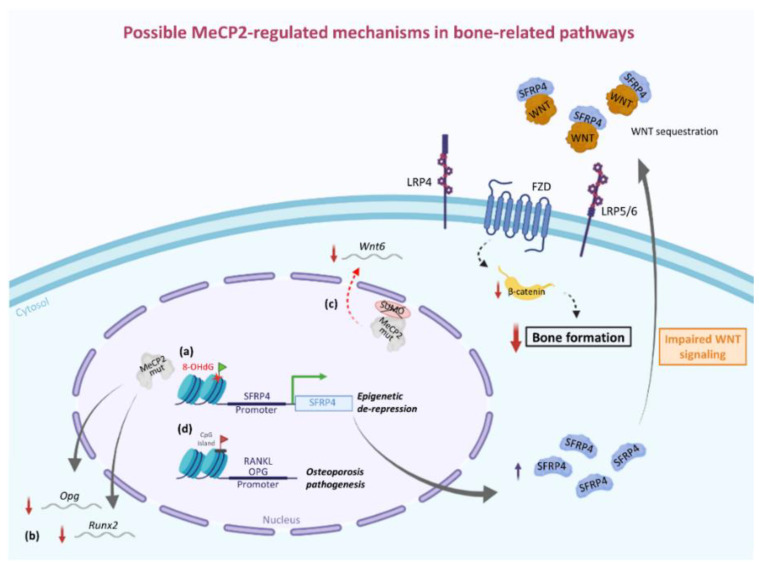
Rett syndrome (RTT) is a monogenic neurodevelopmental disorder primarily caused by mutations in X-linked MECP2 gene, encoding for methyl-CpG binding protein 2 (MeCP2), a multifaceted modulator of gene expression and chromatin organization. Based on the type of mutation, RTT patients exhibit a broad spectrum of clinical phenotypes with various degrees of severity. In addition, as a complex multisystem disease, RTT shows several clinical manifestations ranging from neurological to non-neurological symptoms. The most common non-neurological comorbidities include, among others, orthopedic complications, mainly scoliosis but also early osteopenia/osteoporosis and a high frequency of fractures. A characteristic low bone mineral density dependent on a slow rate of bone formation due to dysfunctional osteoblast activity rather than an increase in bone resorption is at the root of these complications. Evidence from human and animal studies supports the idea that MECP2 mutation could be associated with altered epigenetic regulation of bone-related factors and signaling pathways, including SFRP4/WNT/β-catenin axis and RANKL/RANK/OPG system. More research is needed to better understand the role of MeCP2 in bone homeostasis. Indeed, uncovering the molecular mechanisms underlying RTT bone problems could reveal new potential pharmacological targets for the treatment of these complications that adversely affect the quality of life of RTT patients for whom the only therapeutic approaches currently available include bisphosphonates, dietary supplements, and physical activity.
Keywords: MeCP2; WNT pathway; bone metabolism; bone mineral density; osteoblast.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Leonard H., Bower C., English D. The Prevalence and Incidence of Rett Syndrome in Australia. Eur. Child Adolesc. Psychiatry. 1997;6:8–10. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
