

Bayesian optimization with evolutionary and structure-based regularization for directed protein evolution
- PMID: 34210336
- PMCID: PMC8246133
- DOI: 10.1186/s13015-021-00195-4
Bayesian optimization with evolutionary and structure-based regularization for directed protein evolution
Abstract
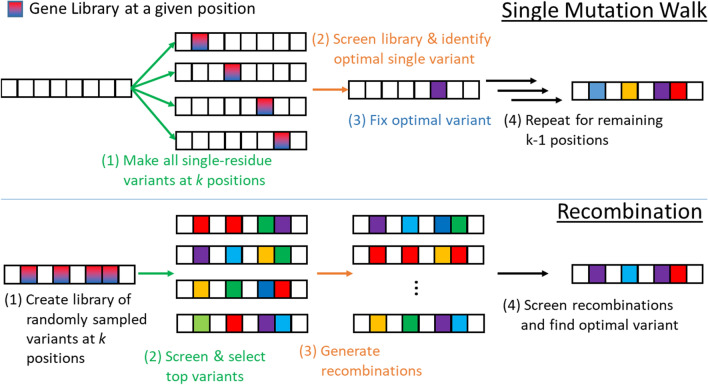
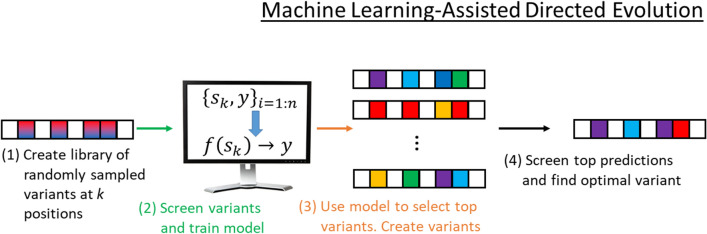
Background: Directed evolution (DE) is a technique for protein engineering that involves iterative rounds of mutagenesis and screening to search for sequences that optimize a given property, such as binding affinity to a specified target. Unfortunately, the underlying optimization problem is under-determined, and so mutations introduced to improve the specified property may come at the expense of unmeasured, but nevertheless important properties (ex. solubility, thermostability, etc). We address this issue by formulating DE as a regularized Bayesian optimization problem where the regularization term reflects evolutionary or structure-based constraints.
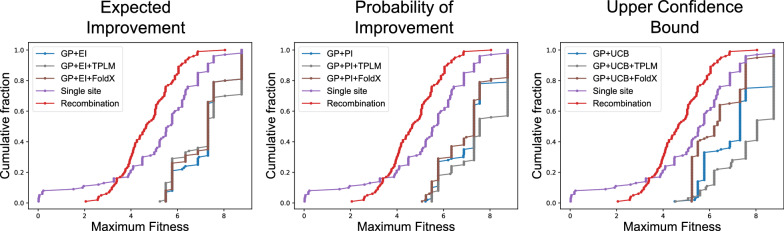
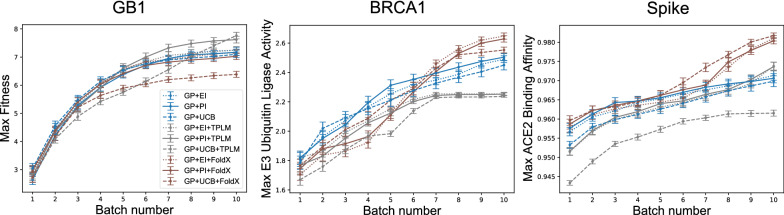
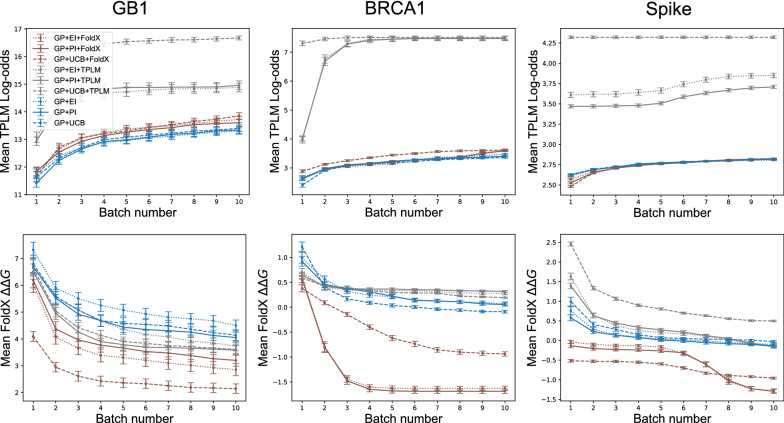
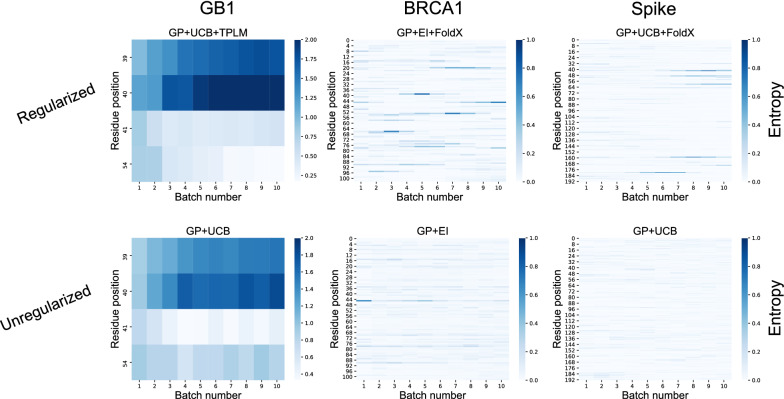
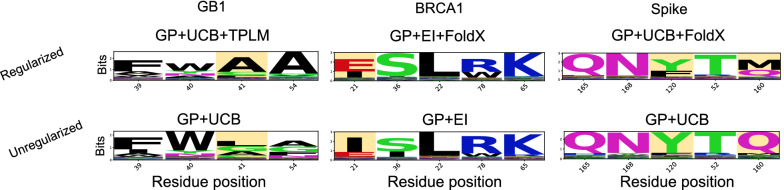
Results: We applied our approach to DE to three representative proteins, GB1, BRCA1, and SARS-CoV-2 Spike, and evaluated both evolutionary and structure-based regularization terms. The results of these experiments demonstrate that: (i) structure-based regularization usually leads to better designs (and never hurts), compared to the unregularized setting; (ii) evolutionary-based regularization tends to be least effective; and (iii) regularization leads to better designs because it effectively focuses the search in certain areas of sequence space, making better use of the experimental budget. Additionally, like previous work in Machine learning assisted DE, we find that our approach significantly reduces the experimental burden of DE, relative to model-free methods.
Conclusion: Introducing regularization into a Bayesian ML-assisted DE framework alters the exploratory patterns of the underlying optimization routine, and can shift variant selections towards those with a range of targeted and desirable properties. In particular, we find that structure-based regularization often improves variant selection compared to unregularized approaches, and never hurts.
Keywords: Active learning; Bayesian optimization; Directed evolution; Gaussian process regression; Protein design; Protein language model; Rational design; Regularization.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Lutz S, Bornscheuer UT. Protein engineering handbook. Weinheim: Wiley-VCH; 2012. OCLC: 890049290.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
