

Editorial
doi: 10.1186/s13059-021-02395-y.
CLIMB-COVID: continuous integration supporting decentralised sequencing for SARS-CoV-2 genomic surveillance
Affiliations
- PMID: 34210356
- PMCID: PMC8247108
- DOI: 10.1186/s13059-021-02395-y
Item in Clipboard
Editorial
CLIMB-COVID: continuous integration supporting decentralised sequencing for SARS-CoV-2 genomic surveillance
Genome Biol.
.
Abstract
In response to the ongoing SARS-CoV-2 pandemic in the UK, the COVID-19 Genomics UK (COG-UK) consortium was formed to rapidly sequence SARS-CoV-2 genomes as part of a national-scale genomic surveillance strategy. The network consists of universities, academic institutes, regional sequencing centres and the four UK Public Health Agencies. We describe the development and deployment of CLIMB-COVID, an encompassing digital infrastructure to address the challenge of collecting and integrating both genomic sequencing data and sample-associated metadata produced across the COG-UK network.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
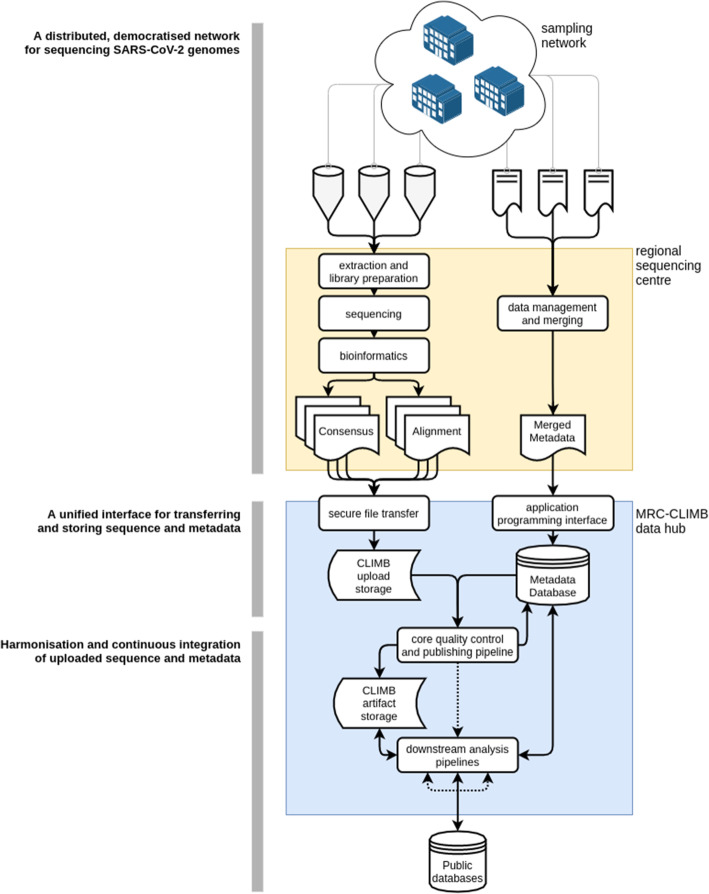
Overview of the COG-UK data flow. (Top) A network of sampling sites (e.g. hospitals and testing centres) produce samples and sample metadata which are received by a regional sequencing centre. The sample is extracted and sequenced and a locally run bioinformatics pipeline generates both a consensus viral genome and an alignment of sequenced read fragments against the SARS-CoV-2 reference genome. (Middle) The consensus sequence and alignments are uploaded via secure file transfer to be stored on CLIMB-COVID. Metadata is securely transferred over HTTPS to an application programming interface (API) that transforms metadata into a model to be stored in a database on CLIMB-COVID. (Bottom) The core quality control pipeline executes every day to integrate newly uploaded samples and metadata into the single canonical dataset of all uploaded sequences. Once this pipeline is finished, it notifies downstream analysis pipelines through a messaging protocol to generate analysis artifacts like phylogenetic trees. Downstream analysis pipelines also automatically deposit genomes in public databases such as GISAID and ENA/INSDC
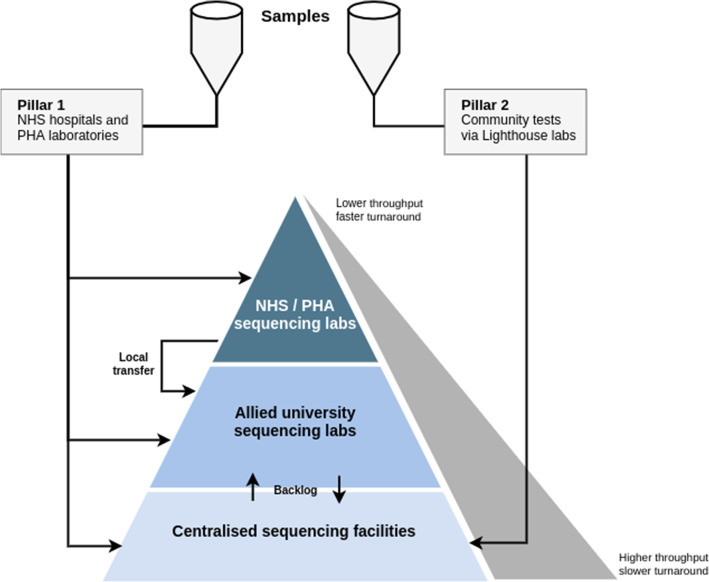
COG-UK sequencing model. Samples are sourced from two “pillars”; pillar 1 samples are collected across the NHS and Public Health Agencies, pillar 2 samples are collected at the Lighthouse Labs at particular strategic sites in the UK. Generally, pillar 1 samples are received by NHS labs who process them for sequencing locally or by a university sequencing lab for a fast turnaround. Pillar 2 samples are generally shipped through to the Wellcome Sanger Institute for high-capacity sequencing
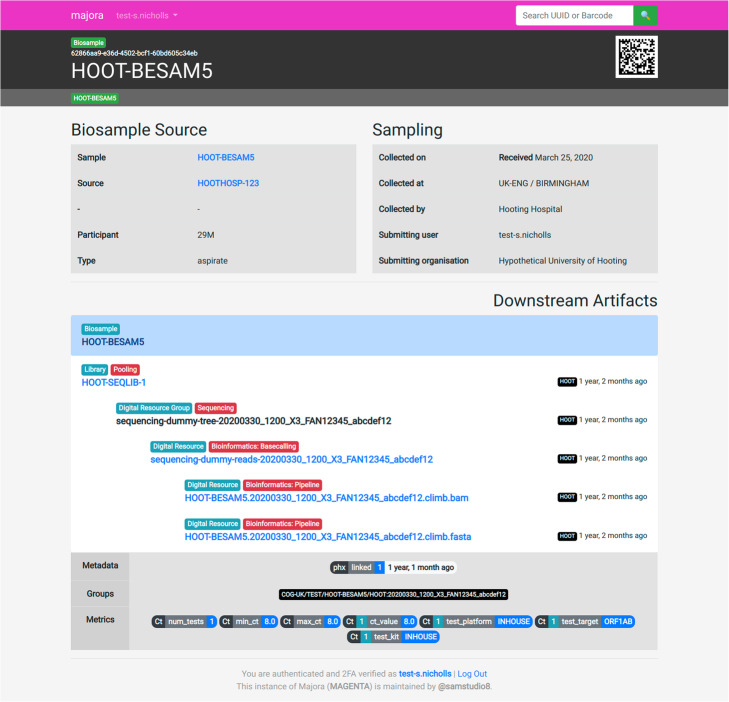
Majora web application biosample view. An example of the web interface presented by Majora to detail a biosample artifact. The downstream artifacts section allows users to see what processes have been applied to the biosample. In this example, the sample was incorporated onto a pooled sequencing library, which was sequenced and basecalled. A downstream bioinformatics pipeline resulted in a FASTA and BAM. Artifacts can be tagged with metadata and metrics. In this example, the artifact is tagged with a linkage flag and information about the sample's cycle threshold value

Example API request to submit a new biosample artifact to Majora. All metadata from biological samples, to library pooling processes and sequencing runs are communicated to Majora through the various API endpoints. These interfaces take structured data in the JSON format and process them to be stored in the Majora’s SQL database. This example demonstrates a simplified request to add a new biosample to Majora (a) and a reply from Majora indicating a validation error (b). Examples rendered with @carbon_app
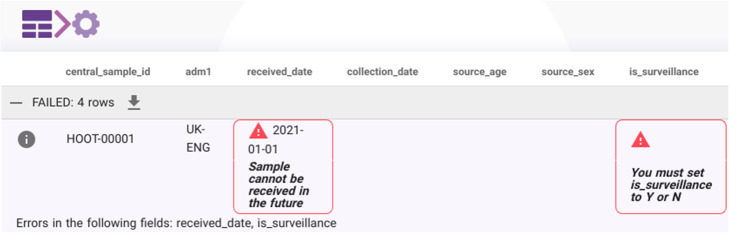
Screenshot of the metadata uploader demonstrating user-facing errors. Metadata is submitted to the consortium by uploading a filled in CSV template to the metadata uploader web application. The uploader converts the CSV data into JSON and communicates with the Majora API. Validation errors are immediately returned, parsed and displayed to the user as shown here
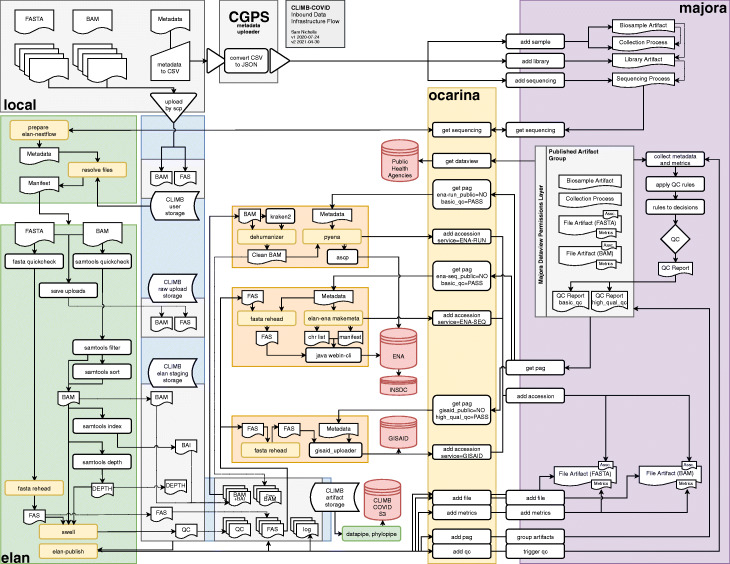
CLIMB-COVID inbound data architecture diagram. Local sites (grey, top left) generate consensus FASTA and alignment BAM for each sequenced sample. Corresponding metadata is collected and managed into a CSV using the consortium template. FASTA and BAM are uploaded to CLIMB using scp or rsync. Metadata is converted from CSV to JSON by the metadata uploader tool and passed to the Majora API to be processed (purple, right). The Elan inbound pipeline (green, left) queries the Majora metadata database using the Ocarina command line client (yellow, right). Elan matches Majora metadata to uploaded files on CLIMB (blue, left) and conducts quality control. Quality metrics are passed to Majora through Ocarina. Outbound distribution pipelines (orange, centre) are able to query Majora using Ocarina and package high-quality sequences for GISAID. ENA and INSDC databases (red). Downstream pipelines (green, centre) such as Datapipe and Phylopipe generate alignments, trees and other analysis artifacts that are shared within the consortium and made publicly available via CLIMB-COVID’s S3 storage (red). Programmes in yellow boxes indicate software built specifically for CLIMB-COVID
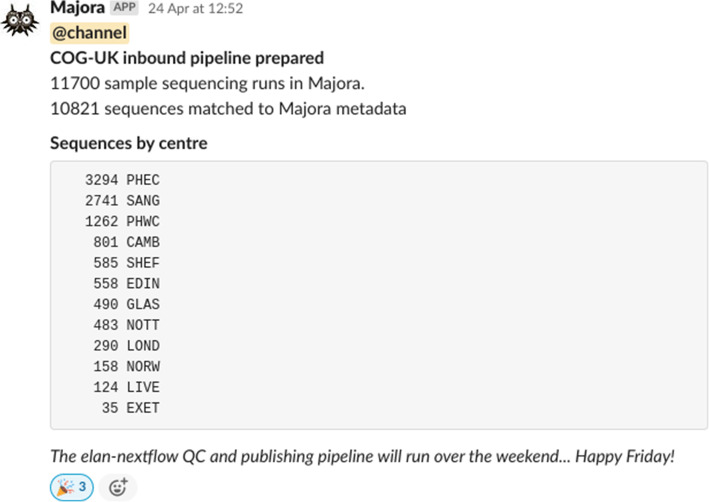
An automated Slack message announcing the start of Elan pipeline. The Elan inbound distribution pipeline is operated transparently by providing a series of courtesy messages before and after it has run. Slack messages are sent programmatically through a web hook to announce samples that appear to be missing metadata or a genome sequence. This example, dated April 24, 2020, announces that Elan was about to process the 10,000th sample
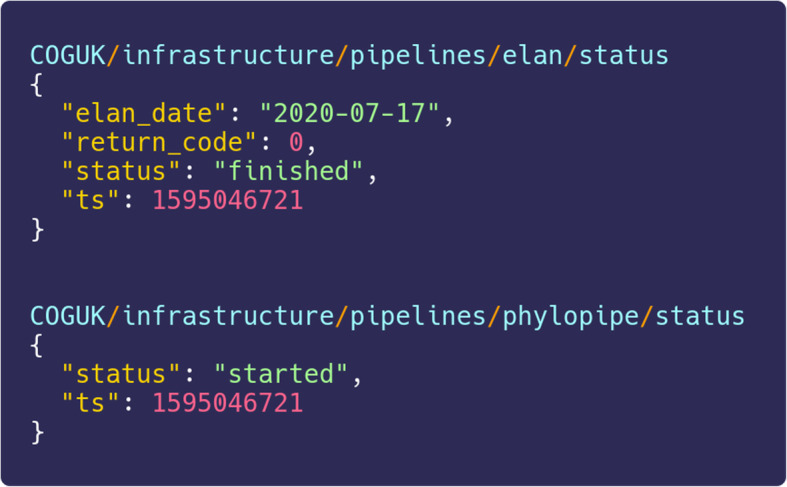
Automated machine-readable messages exchanged between pipelines from a messaging queue. To assist orchestration of pipelines, we run a message broker service that allows different pipelines within COG-UK to send messages and interact with each other. This example shows Elan’s first message on July 17, 2020, emitted to announce it has successfully completed, and the phylogenetics pipeline responding to say it has started as a result of the new data to be processed

Example tree from Grapevine. A phylogenetic tree of COG-UK sequences (coloured) against a background subset of non-UK sequences from GISAID (grey) from the last 100 days, produced by Grapevine. All UK sequences are coloured by the week in which they were sampled

Example Civet report based on a simulated outbreak. The customisable preamble can contain information such as a description of the outbreak, number of sequences of interest and authors of the report (A). The report also includes summary tables of the input data, split by whether the queries are in the COG-UK dataset or have been provided by the user (B). Civet displays summarised subtrees of the local phylogenetic diversity surrounding sequences of interest, with tips coloured by administrative level one region by default (C). Optionally, a timeline of the sequences can be displayed (D), and a ‘Snipit graph’ which highlights nucleotide changes from a defined reference genome sequence amongst the sequences of interest (E). Descriptive maps show the geographic distribution and the genetic diversity of SARS-CoV-2 circulating in the local area (F)
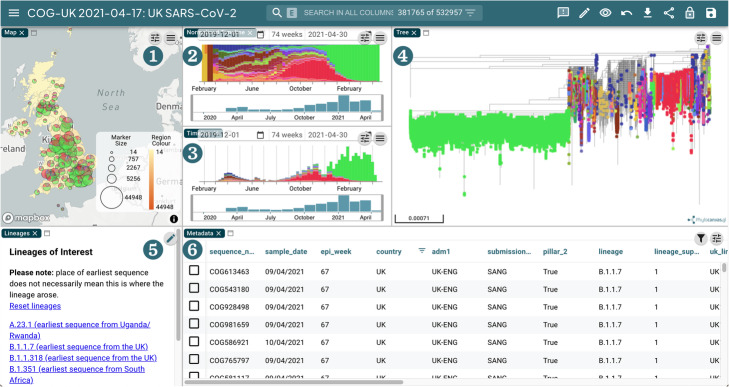
Screenshot of the COG-UK Microreact instance. 1 A map view showing the place of sample collection. 2, 3 Normalised and standard timelines showing the proportion and number of each lineage found in the samples sequenced over time respectively. 4 A phylogeny derived from the analysis described in the section above. 5 Panel allowing quick filtering by lineages of interest. 6 A metadata table view allowing filtering and sorting of data. These views are generated with COG-UK data that has been processed by Elan and the phylogenetics pipeline
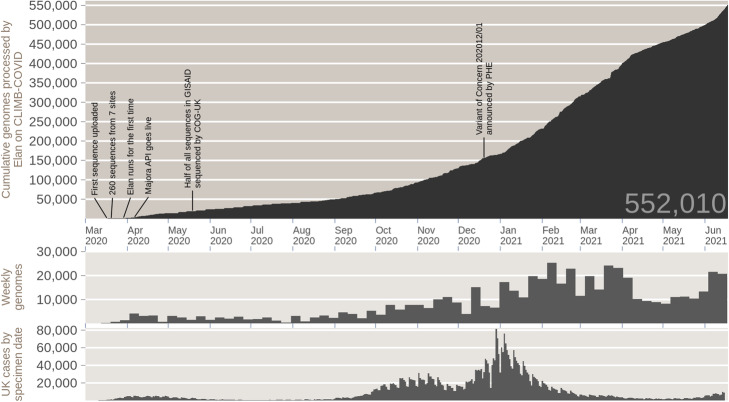
Cumulative total sequences and weekly sequences processed by Elan on CLIMB-COVID, compared to UK daily SARS-CoV-2 positive cases. (Top) Cumulative total genomes processed by Elan on CLIMB-COVID since March 2020. (Middle) Number of genomes processed per week by Elan on CLIMB-COVID. (Bottom) Daily positive SARS-CoV-2 cases in the UK (by specimen date) as reported by the UK government website for data and insights on coronavirus (coronavirus.data.gov.uk )
References
-
- Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, Ouédraogo N, Afrough B, Bah A, Baum JHJ, Becker-Ziaja B, Boettcher JP, Cabeza-Cabrerizo M, Camino-Sánchez Á, Carter LL, Doerrbecker J, Enkirch T, Dorival IG, Hetzelt N, Hinzmann J, Holm T, Kafetzopoulou LE, Koropogui M, Kosgey A, Kuisma E, Logue CH, Mazzarelli A, Meisel S, Mertens M, Michel J, Ngabo D, Nitzsche K, Pallasch E, Patrono LV, Portmann J, Repits JG, Rickett NY, Sachse A, Singethan K, Vitoriano I, Yemanaberhan RL, Zekeng EG, Racine T, Bello A, Sall AA, Faye O, Faye O, Magassouba N’F, Williams CV, Amburgey V, Winona L, Davis E, Gerlach J, Washington F, Monteil V, Jourdain M, Bererd M, Camara A, Somlare H, Camara A, Gerard M, Bado G, Baillet B, Delaune D, Nebie KY, Diarra A, Savane Y, Pallawo RB, Gutierrez GJ, Milhano N, Roger I, Williams CJ, Yattara F, Lewandowski K, Taylor J, Rachwal P, J. Turner D, Pollakis G, Hiscox JA, Matthews DA, Shea MKO’, Johnston AMD, Wilson D, Hutley E, Smit E, di Caro A, Wölfel R, Stoecker K, Fleischmann E, Gabriel M, Weller SA, Koivogui L, Diallo B, Keïta S, Rambaut A, Formenty P, Günther S, Carroll MW. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228–232. doi: 10.1038/nature16996. - DOI - PMC - PubMed
-
- Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
