

A fluorescence-based genetic screen reveals diverse mechanisms silencing small RNA signaling in E. coli
- PMID: 34210798
- PMCID: PMC8271630
- DOI: 10.1073/pnas.2106964118
A fluorescence-based genetic screen reveals diverse mechanisms silencing small RNA signaling in E. coli
Abstract
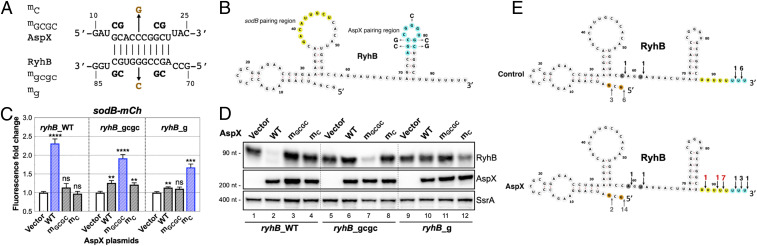
As key players of gene regulation in many bacteria, small regulatory RNAs (sRNAs) associated with the RNA chaperone Hfq shape numerous phenotypic traits, including metabolism, stress response and adaptation, as well as virulence. sRNAs can alter target messenger RNA (mRNA) translation and stability via base pairing. sRNA synthesis is generally under tight transcriptional regulation, but other levels of regulation of sRNA signaling are less well understood. Here we used a fluorescence-based functional screen to identify regulators that can quench sRNA signaling of the iron-responsive sRNA RyhB in Escherichia coli The identified regulators fell into two classes, general regulators (affecting signaling by many sRNAs) and RyhB-specific regulators; we focused on the specific ones here. General regulators include three Hfq-interacting sRNAs, CyaR, ChiX, and McaS, previously found to act through Hfq competition, RNase T, a 3' to 5' exonuclease not previously implicated in sRNA degradation, and YhbS, a putative GCN5-related N-acetyltransferase (GNAT). Two specific regulators were identified. AspX, a 3'end-derived small RNA, specifically represses RyhB signaling via an RNA sponging mechanism. YicC, a previously uncharacterized but widely conserved protein, triggers rapid RyhB degradation via collaboration with the exoribonuclease PNPase. These findings greatly expand our knowledge of regulation of bacterial sRNA signaling and suggest complex regulatory networks for controlling iron homeostasis in bacteria. The fluorescence-based genetic screen system described here is a powerful tool expected to accelerate the discovery of novel regulators of sRNA signaling in many bacteria.
Keywords: Hfq competition; RNA chaperone; RNA sponge; RyhB; exoribonuclease.
Conflict of interest statement
The authors declare no competing interest.
Figures







References
-
- Wagner E. G. H., Romby P., Small RNAs in bacteria and archaea: Who they are, what they do, and how they do it. Adv. Genet. 90, 133–208 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
