

Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant
- PMID: 34210996
- PMCID: PMC8249556
- DOI: 10.1038/s41598-021-91662-w
Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant
Abstract
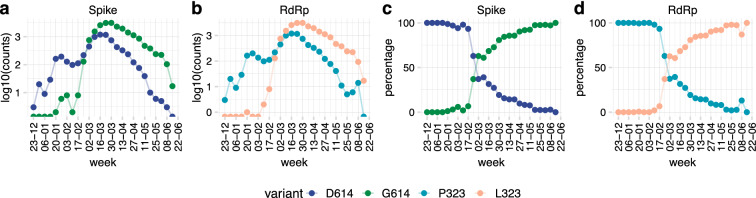
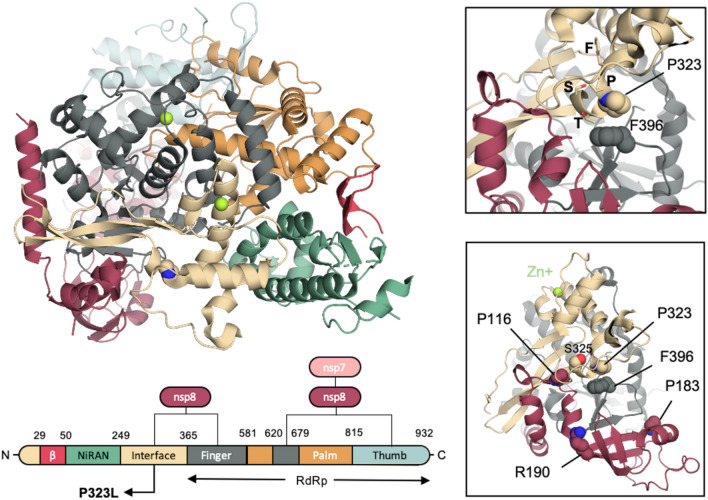
The D614G mutation in the Spike protein of the SARS-CoV-2 has effectively replaced the early pandemic-causing variant. Using pseudotyped lentivectors, we confirmed that the aspartate replacement by glycine in position 614 is markedly more infectious. Molecular modelling suggests that the G614 mutation facilitates transition towards an open state of the Spike protein. To explain the epidemiological success of D614G, we analysed the evolution of 27,086 high-quality SARS-CoV-2 genome sequences from GISAID. We observed striking coevolution of D614G with the P323L mutation in the viral polymerase. Importantly, the exclusive presence of G614 or L323 did not become epidemiologically relevant. In contrast, the combination of the two mutations gave rise to a viral G/L variant that has all but replaced the initial D/P variant. Our results suggest that the P323L mutation, located in the interface domain of the RNA-dependent RNA polymerase, is a necessary alteration that led to the epidemiological success of the present variant of SARS-CoV-2. However, we did not observe a significant correlation between reported COVID-19 mortality in different countries and the prevalence of the Wuhan versus G/L variant. Nevertheless, when comparing the speed of emergence and the ultimate predominance in individual countries, it is clear that the G/L variant displays major epidemiological supremacy over the original variant.
Conflict of interest statement
MC worked for Neurix SA. All authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
