

High-precision and cost-efficient sequencing for real-time COVID-19 surveillance
- PMID: 34211026
- PMCID: PMC8249533
- DOI: 10.1038/s41598-021-93145-4
High-precision and cost-efficient sequencing for real-time COVID-19 surveillance
Abstract
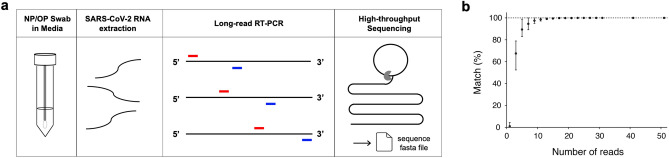
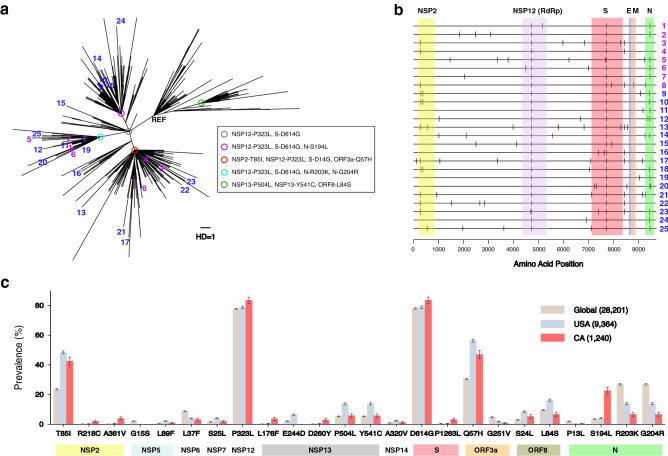
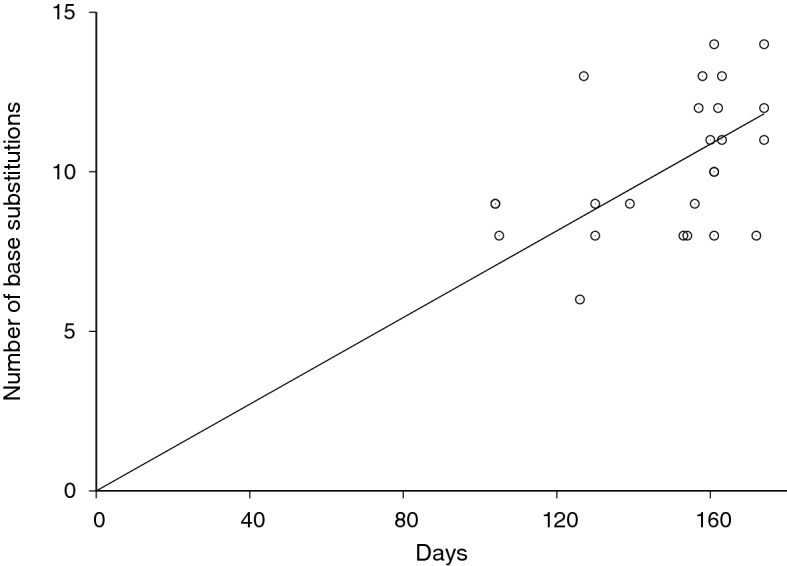
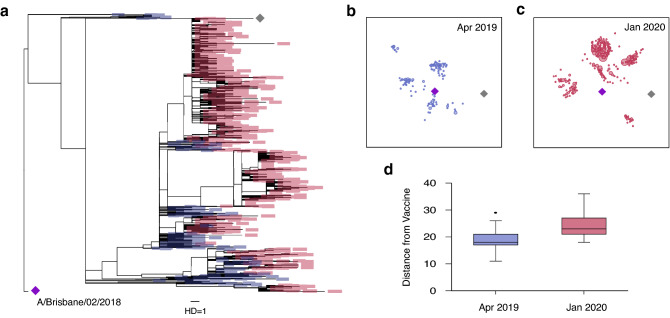
COVID-19 global cases have climbed to more than 33 million, with over a million total deaths, as of September, 2020. Real-time massive SARS-CoV-2 whole genome sequencing is key to tracking chains of transmission and estimating the origin of disease outbreaks. Yet no methods have simultaneously achieved high precision, simple workflow, and low cost. We developed a high-precision, cost-efficient SARS-CoV-2 whole genome sequencing platform for COVID-19 genomic surveillance, CorvGenSurv (Coronavirus Genomic Surveillance). CorvGenSurv directly amplified viral RNA from COVID-19 patients' Nasopharyngeal/Oropharyngeal (NP/OP) swab specimens and sequenced the SARS-CoV-2 whole genome in three segments by long-read, high-throughput sequencing. Sequencing of the whole genome in three segments significantly reduced sequencing data waste, thereby preventing dropouts in genome coverage. We validated the precision of our pipeline by both control genomic RNA sequencing and Sanger sequencing. We produced near full-length whole genome sequences from individuals who were COVID-19 test positive during April to June 2020 in Los Angeles County, California, USA. These sequences were highly diverse in the G clade with nine novel amino acid mutations including NSP12-M755I and ORF8-V117F. With its readily adaptable design, CorvGenSurv grants wide access to genomic surveillance, permitting immediate public health response to sudden threats.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
