

Using X-ray free-electron lasers for spectroscopy of molecular catalysts and metalloenzymes
- PMID: 34212130
- PMCID: PMC8245202
- DOI: 10.1038/s42254-021-00289-3
Using X-ray free-electron lasers for spectroscopy of molecular catalysts and metalloenzymes
Abstract
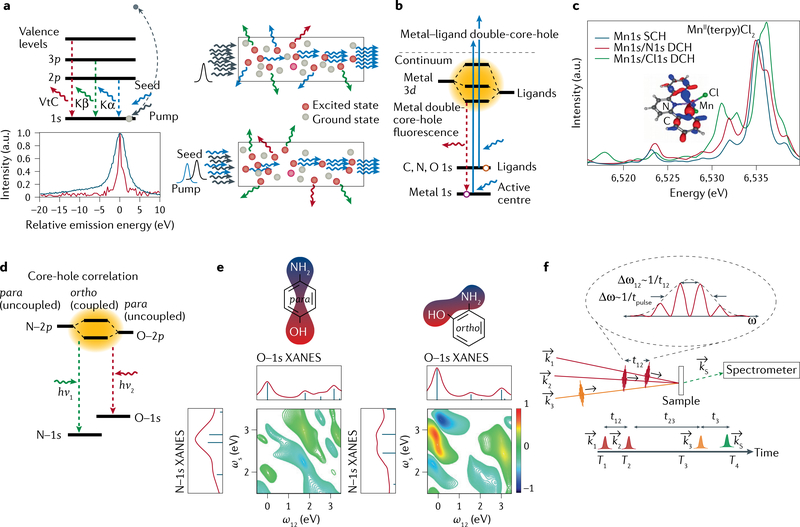
The metal centres in metalloenzymes and molecular catalysts are responsible for the rearrangement of atoms and electrons during complex chemical reactions, and they enable selective pathways of charge and spin transfer, bond breaking/making and the formation of new molecules. Mapping the electronic structural changes at the metal sites during the reactions gives a unique mechanistic insight that has been difficult to obtain to date. The development of X-ray free-electron lasers (XFELs) enables powerful new probes of electronic structure dynamics to advance our understanding of metalloenzymes. The ultrashort, intense and tunable XFEL pulses enable X-ray spectroscopic studies of metalloenzymes, molecular catalysts and chemical reactions, under functional conditions and in real time. In this Technical Review, we describe the current state of the art of X-ray spectroscopy studies at XFELs and highlight some new techniques currently under development. With more XFEL facilities starting operation and more in the planning or construction phase, new capabilities are expected, including high repetition rate, better XFEL pulse control and advanced instrumentation. For the first time, it will be possible to make real-time molecular movies of metalloenzymes and catalysts in solution, while chemical reactions are taking place.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
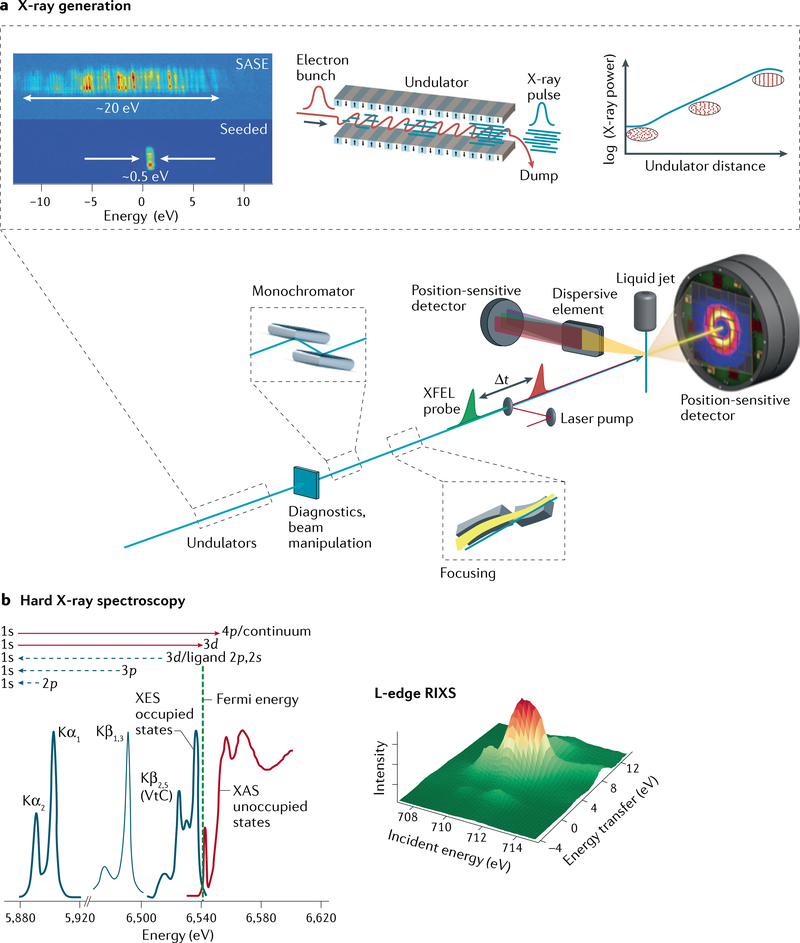
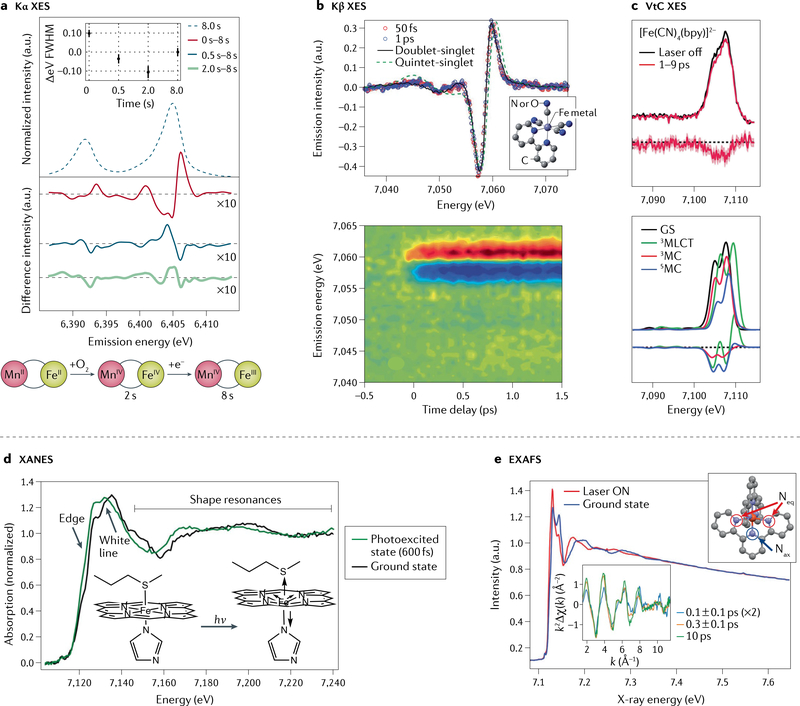
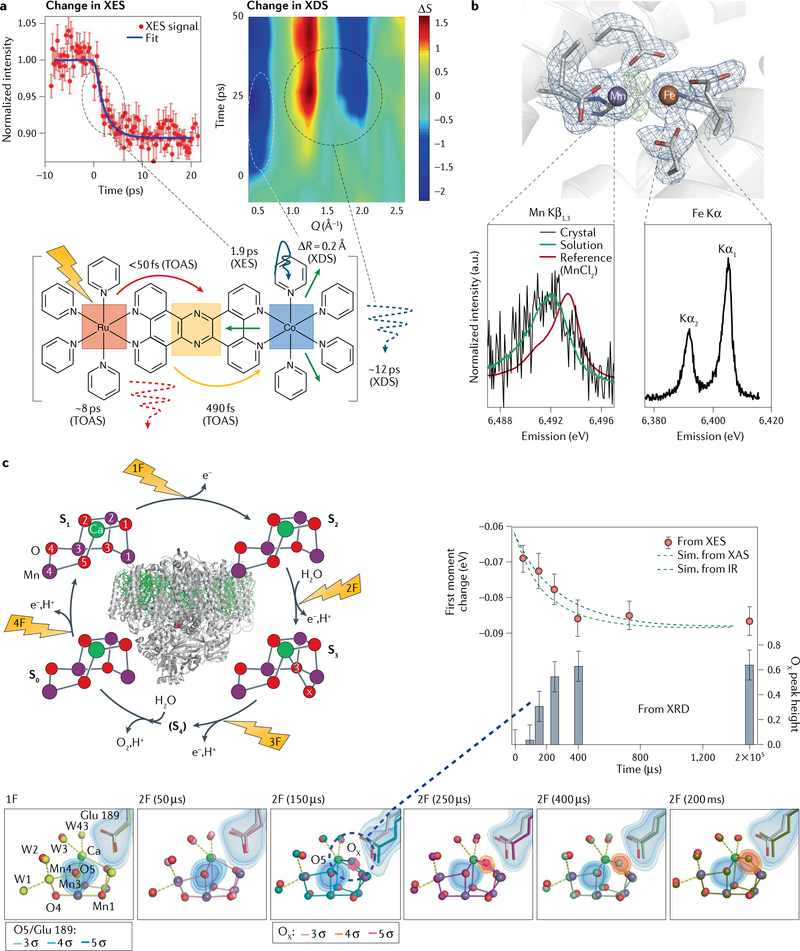
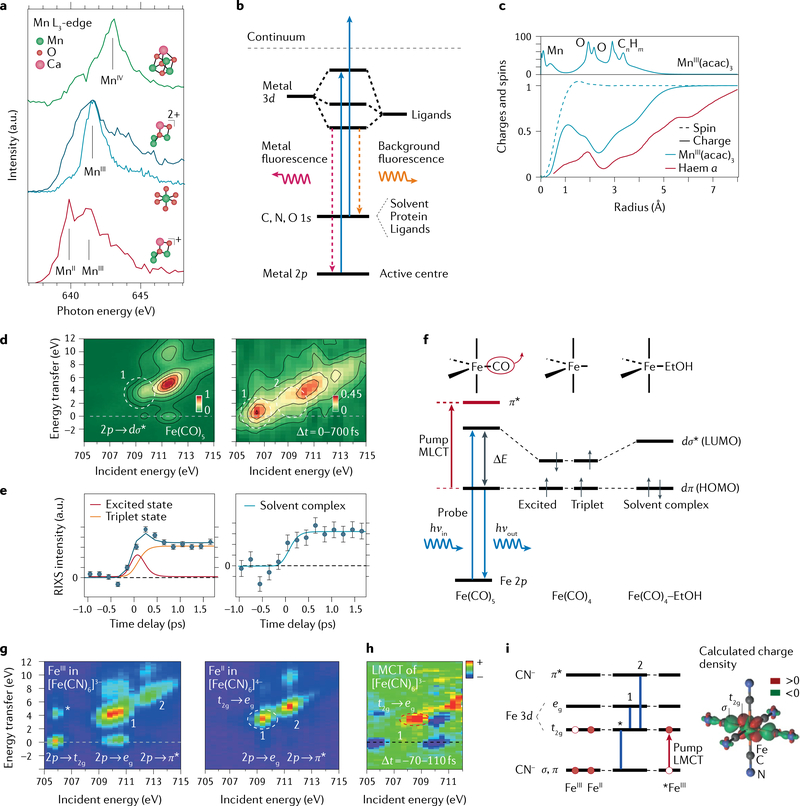
References
-
- Röntgen WC On a new kind of rays. Science 3, 227–231 (1896). - PubMed
-
- Dam HJW in McClure’s Magazine Vol. 6 (McClure SS, 1896).
-
- Watson JD & Crick FHC Molecular structure of nucleic acids - a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953). - PubMed
-
- Rossbach J, Schneider JR & Wurth W 10 years of pioneering X-ray science at the free-electron laser FLASH at DESY. Phys. Rep 808, 1–74 (2019).