

Kratom Alkaloids as Probes for Opioid Receptor Function: Pharmacological Characterization of Minor Indole and Oxindole Alkaloids from Kratom
- PMID: 34213886
- PMCID: PMC8328003
- DOI: 10.1021/acschemneuro.1c00149
Kratom Alkaloids as Probes for Opioid Receptor Function: Pharmacological Characterization of Minor Indole and Oxindole Alkaloids from Kratom
Abstract
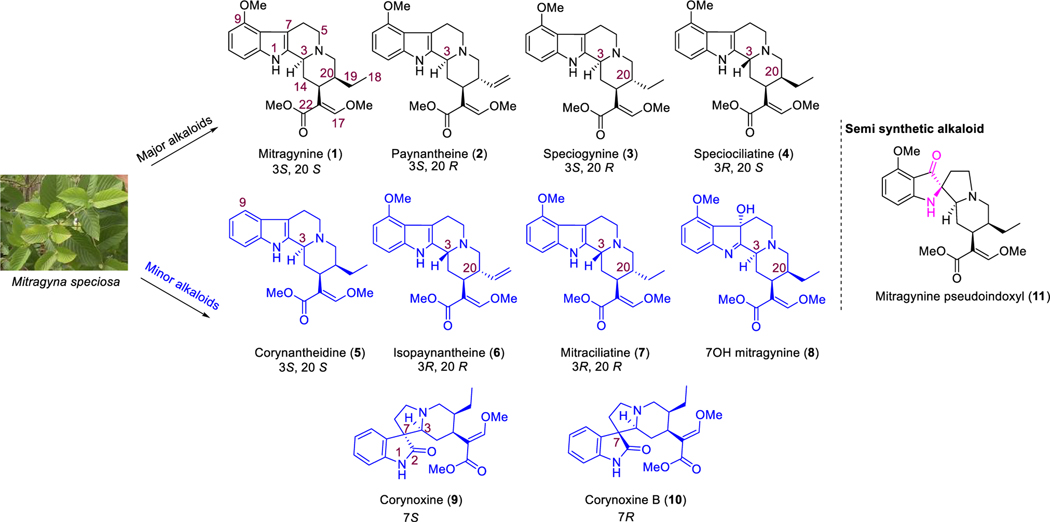
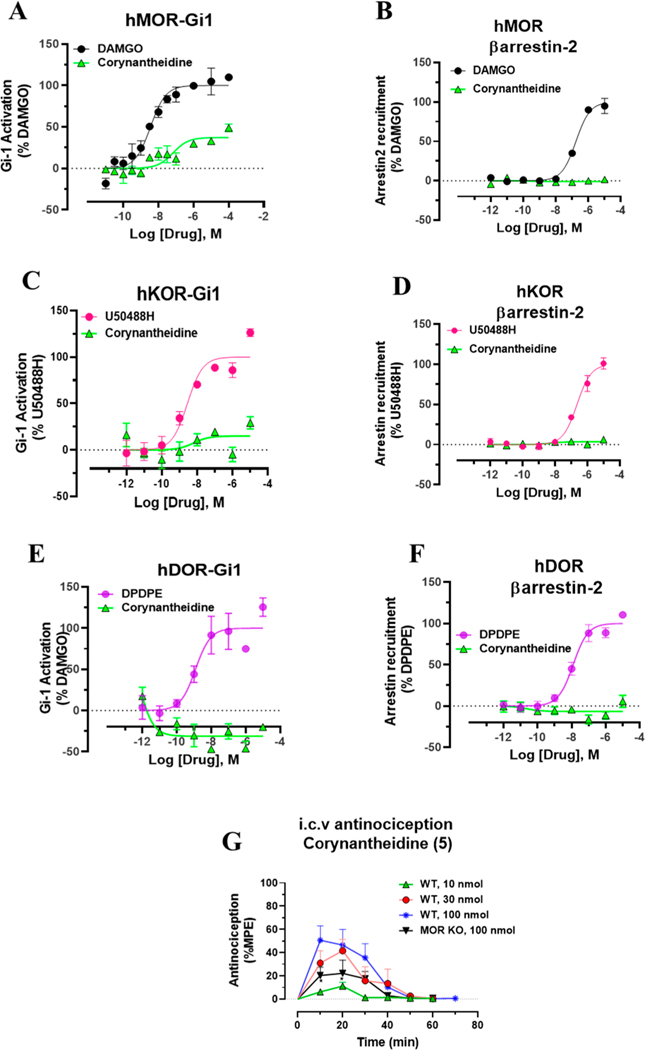
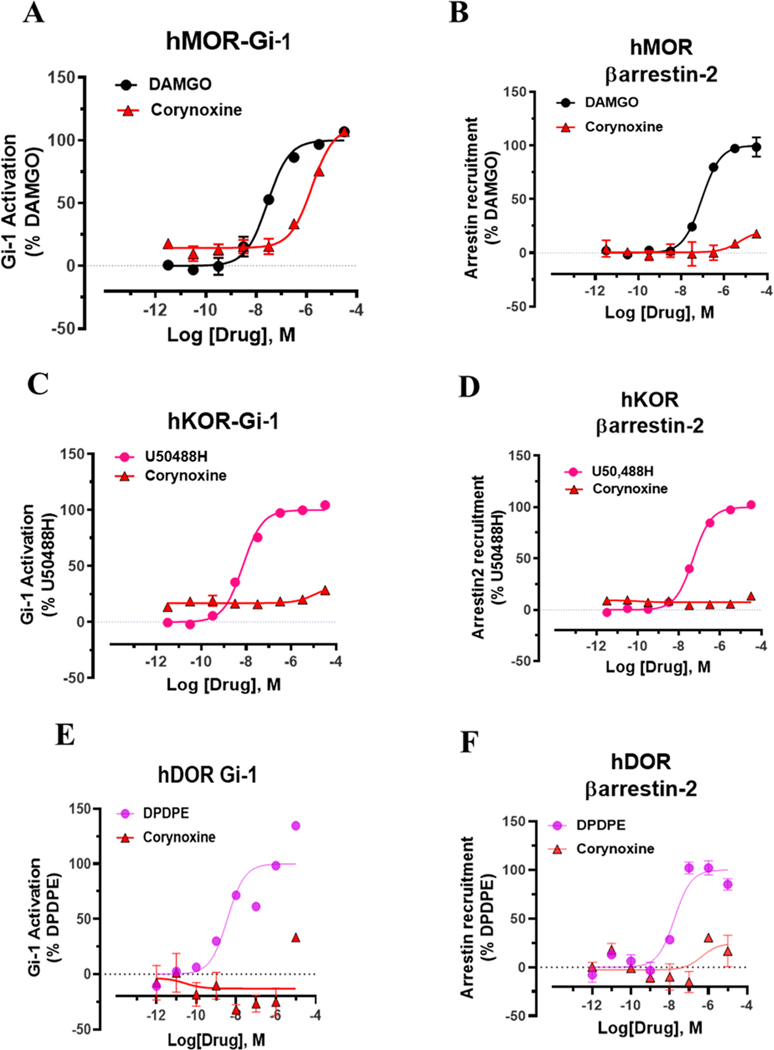
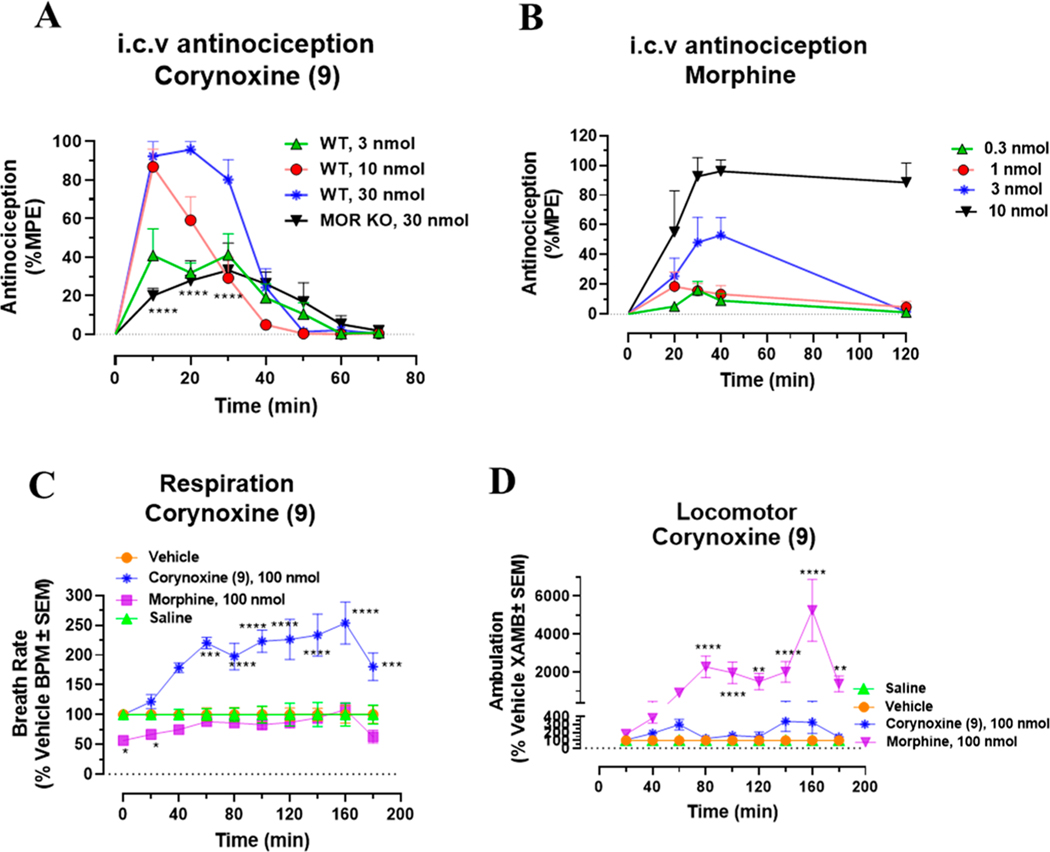
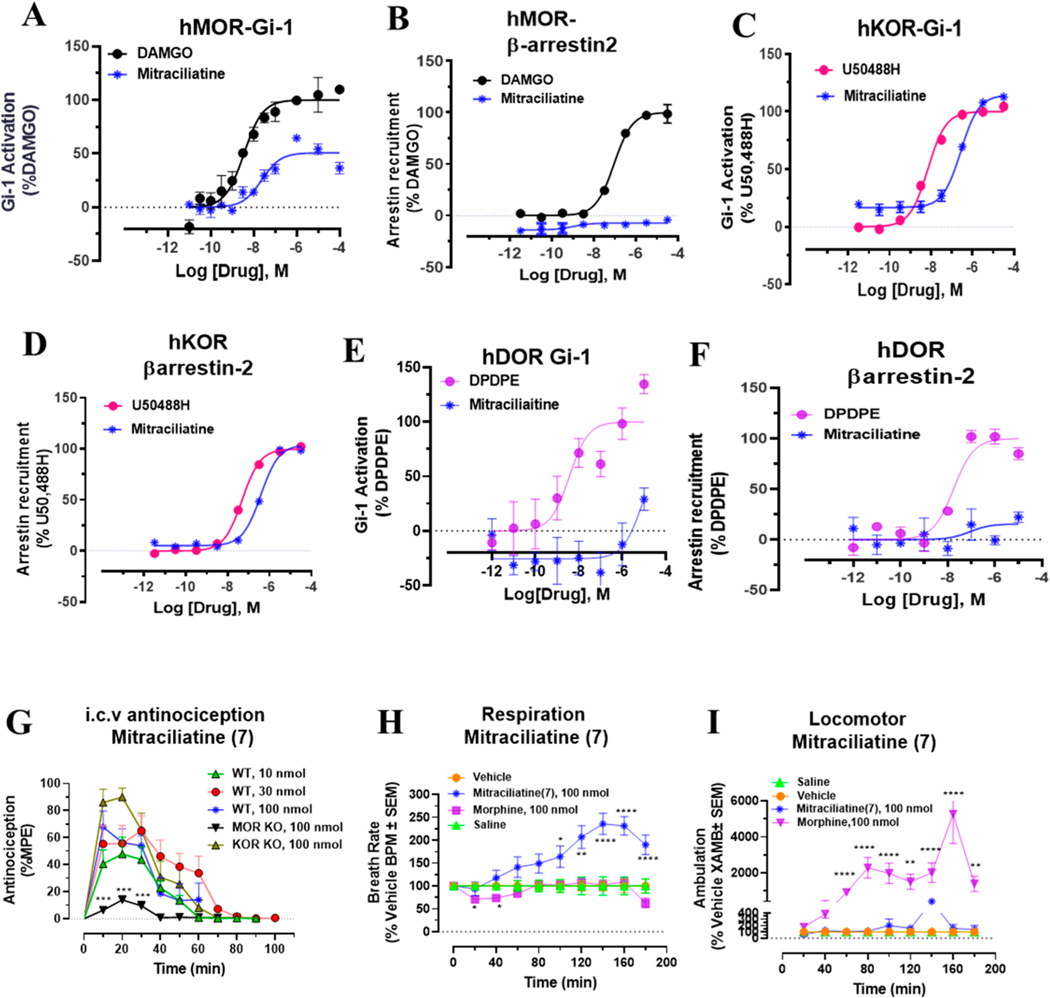
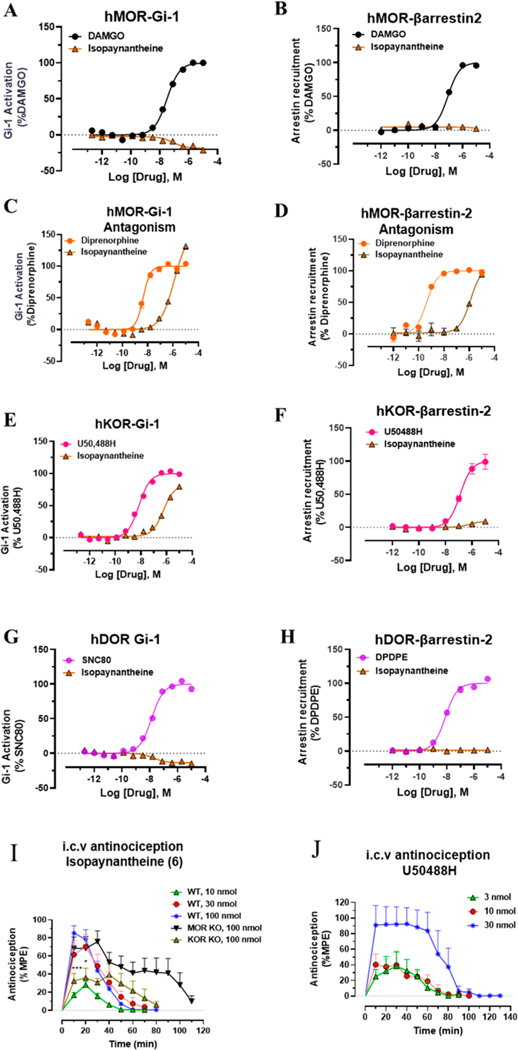
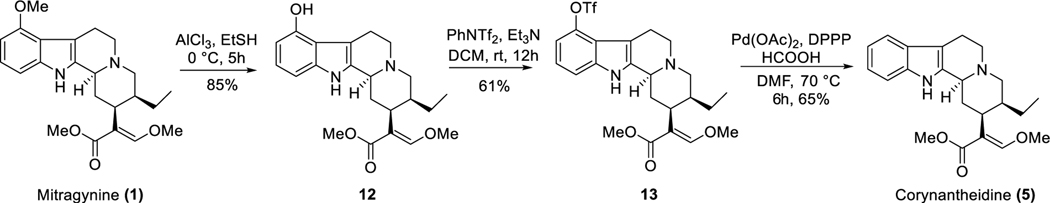
Dry leaves of kratom (mitragyna speciosa) are anecdotally consumed as pain relievers and antidotes against opioid withdrawal and alcohol use disorders. There are at least 54 alkaloids in kratom; however, investigations to date have focused around mitragynine, 7-hydroxy mitragynine (7OH), and mitragynine pseudoindoxyl (MP). Herein, we probe a few minor indole and oxindole based alkaloids, reporting the receptor affinity, G-protein activity, and βarrestin-2 signaling of corynantheidine, corynoxine, corynoxine B, mitraciliatine, and isopaynantheine at mouse and human opioid receptors. We identify corynantheidine as a mu opioid receptor (MOR) partial agonist, whereas its oxindole derivative corynoxine was an MOR full agonist. Similarly, another alkaloid mitraciliatine was found to be an MOR partial agonist, while isopaynantheine was a KOR agonist which showed reduced βarrestin-2 recruitment. Corynantheidine, corynoxine, and mitraciliatine showed MOR dependent antinociception in mice, but mitraciliatine and corynoxine displayed attenuated respiratory depression and hyperlocomotion compared to the prototypic MOR agonist morphine in vivo when administered supraspinally. Isopaynantheine on the other hand was identified as the first kratom derived KOR agonist in vivo. While these minor alkaloids are unlikely to play the majority role in the biological actions of kratom, they represent excellent starting points for further diversification as well as distinct efficacy and signaling profiles with which to probe opioid actions in vivo.
Keywords: Respiration; corynoxine; kratom; mitraciliatine; oxindoles; partial agonism.
Conflict of interest statement
The authors declare the following competing financial interest(s): S.M. is a co-founder of Sparian Inc. S.M. is an inventor on patent applications related to mitragynine analogues, which may lead to royalties or other licensing revenues from future commercial products.
Figures
References
-
- Compton WM, Jones CM, and Baldwin GT (2016) The Authors Reply. New England Journal of Medicine, 1296. - PubMed
-
- Overdose Death Rates, National Institute on Drug Abuse (NIDA), https://www.drugabuse.gov/related-topics/trendsstatistics/overdose-death... (accessed 2019-09-23).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
