

Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma
- PMID: 34215733
- PMCID: PMC8253809
- DOI: 10.1038/s41467-021-24168-8
Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma
Abstract
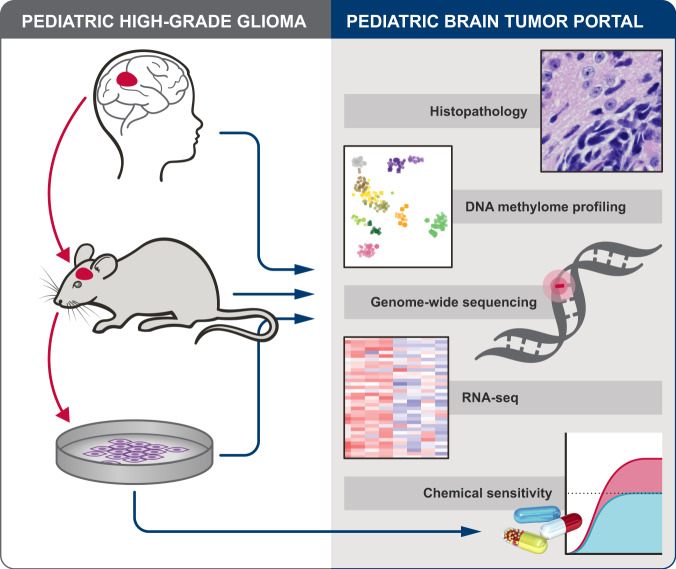
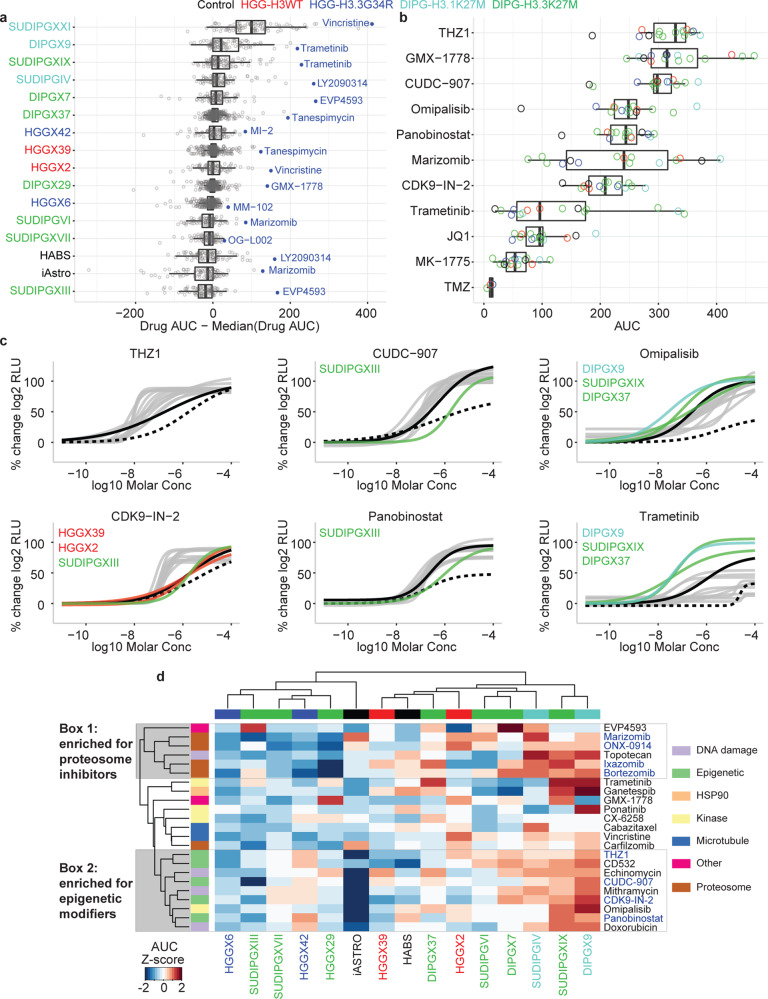
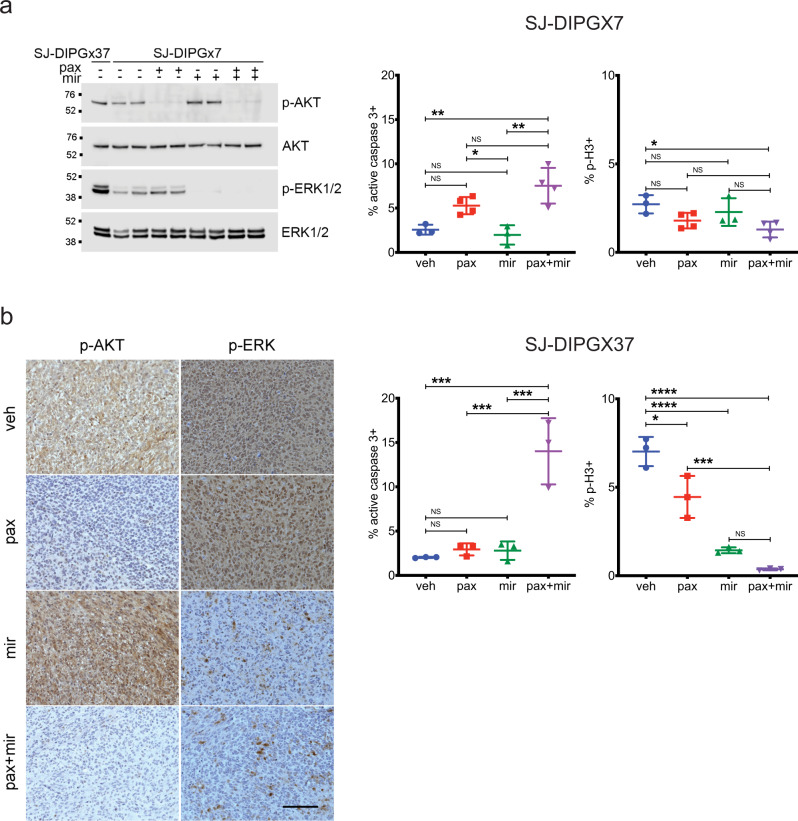
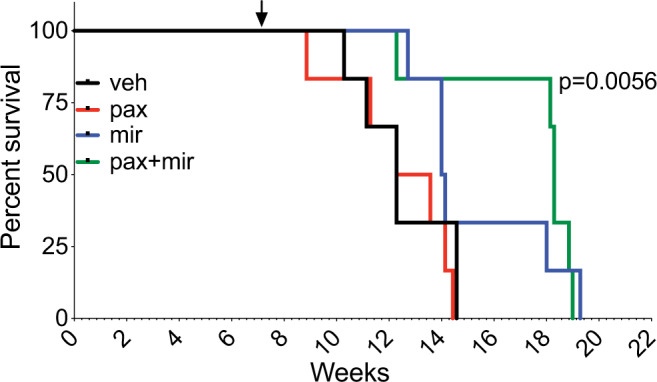
Pediatric high-grade glioma (pHGG) is a major contributor to cancer-related death in children. In vitro and in vivo disease models reflecting the intimate connection between developmental context and pathogenesis of pHGG are essential to advance understanding and identify therapeutic vulnerabilities. Here we report establishment of 21 patient-derived pHGG orthotopic xenograft (PDOX) models and eight matched cell lines from diverse groups of pHGG. These models recapitulate histopathology, DNA methylation signatures, mutations and gene expression patterns of the patient tumors from which they were derived, and include rare subgroups not well-represented by existing models. We deploy 16 new and existing cell lines for high-throughput screening (HTS). In vitro HTS results predict variable in vivo response to PI3K/mTOR and MEK pathway inhibitors. These unique new models and an online interactive data portal for exploration of associated detailed molecular characterization and HTS chemical sensitivity data provide a rich resource for pediatric brain tumor research.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
