

Identification of a Novel Splice Site Mutation in RUNX2 Gene in a Family with Rare Autosomal Dominant Cleidocranial Dysplasia
- PMID: 34217160
- PMCID: PMC8334394
- DOI: 10.52547/ibj.25.4.297
Identification of a Novel Splice Site Mutation in RUNX2 Gene in a Family with Rare Autosomal Dominant Cleidocranial Dysplasia
Abstract
Background: Pathogenic variants of RUNX2, a gene that encodes an osteoblast-specific transcription factor, have been shown as the cause of Cleidocranial dysplasia (CCD), which is a rare hereditary skeletal and dental disorder with dominant mode of inheritance and a broad range of clinical variability. Due to the relative lack of clinical complications resulting in CCD, the medical diagnosis of this disorder is challenging, which leaves it underdiagnosed.
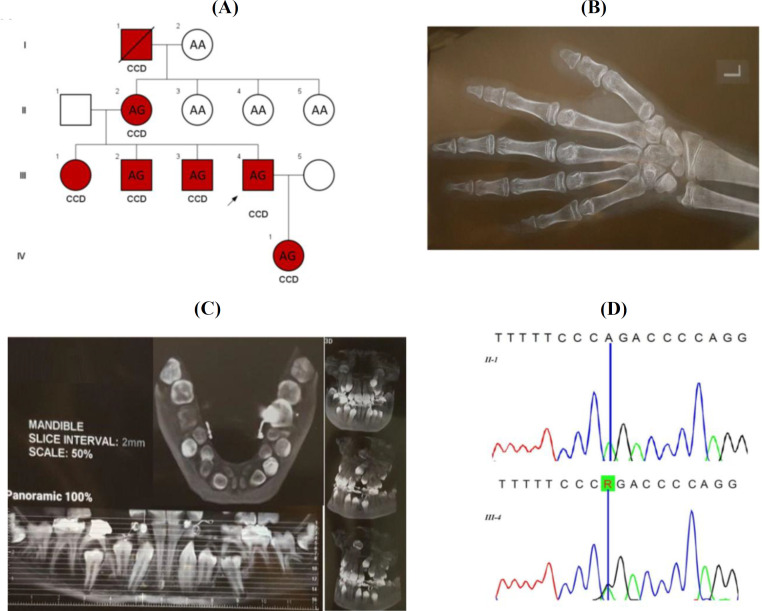
Methods: : In this study, nine healthy and affected members of an Iranian family were investigated. PCR and sequencing of all exons and exon-intron boundaries of runt-related transcription factor 2 (RUNX2; NM_001024630) gene was performed on proband. Co-segregation analysis was conducted in the other family members for the identified variant. Additionally, a cohort of 100 Iranian ethnicity-matched healthy controls was screened by Amplification Refractory Mutation System PCR method.
Results: The novel splice site variant (c.860-2A>G), which was identified in the intron 6 of RUNX2 gene, co-segregated with the disease in the family, and it was absent in healthy controls. Pathogenicity of this variant was determined by several software, including , human splicing finder, which predicts the formation or disruption of splice donor sites, splice acceptor sites, exonic splicing silencer sites, and exonic splicing enhancer sites. In silico analysis predicted this novel variant to be disease causing.
Conclusion: The identified variant is predicted to have an effect on splicing, which leads to exon skipping and producing a truncated protein via introducing a premature stop codon.
Keywords: Cleidocranial dysplasia; RUNX2; Splice site.
Conflict of interest statement
None declared.
Figures
References
-
- Marie P, Sainton P. Sur la dysostose cleido-clanienne hereditaire. Revista de Neurología. 1898;6:835–838.
-
- Keats TE. Cleidocranial dysostosis: some atypical roentgen manifestations. The American journal of roentgenology radium therapy and nuclear medicine. 1967;100(1):71–74. - PubMed
-
- Jarvis JL, KEATS TE. Cleidocranial dysostosis: A review of 40 new cases. American journal of roentgenology. 1974;121(1):5–16. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources