

Evolution of Our Understanding of XIAP Deficiency
- PMID: 34222142
- PMCID: PMC8247594
- DOI: 10.3389/fped.2021.660520
Evolution of Our Understanding of XIAP Deficiency
Abstract
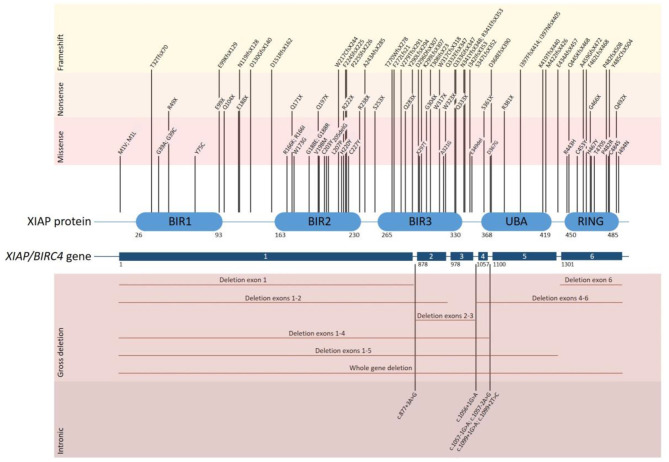
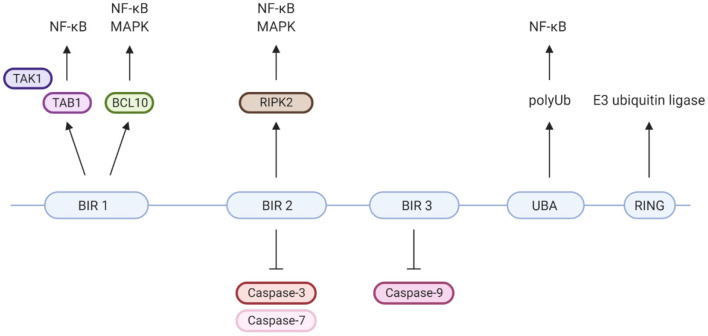
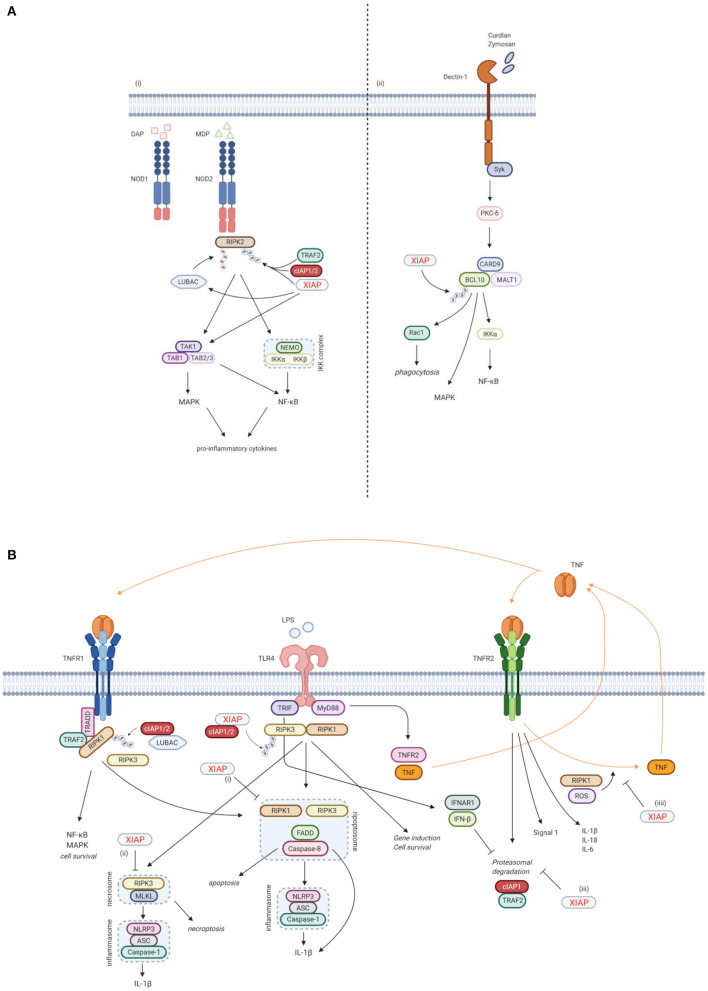
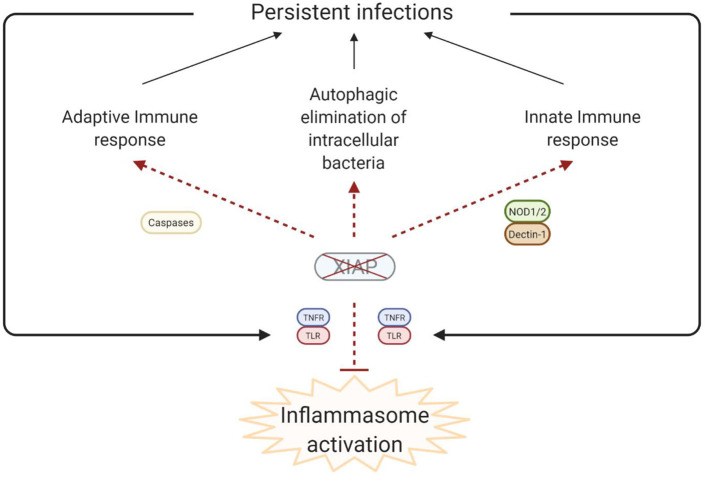
X-linked inhibitor of apoptosis (XIAP) deficiency is a rare inborn error of immunity first described in 2006. XIAP deficiency is characterised by immune dysregulation and a broad spectrum of clinical manifestations, including haemophagocytic lymphohistiocytosis (HLH), inflammatory bowel disease (IBD), hypogammaglobulinemia, susceptibility to infections, splenomegaly, cytopaenias, and other less common autoinflammatory phenomena. Since the first description of the disease, many XIAP deficient patients have been identified and our understanding of the disease has grown. Over 90 disease causing mutations have been described and more inflammatory disease manifestations, such as hepatitis, arthritis, and uveitis, are now well-recognised. Recently, following the introduction of reduced intensity conditioning (RIC), outcomes of allogeneic haematopoietic stem cell transplantation (HSCT), the only curative treatment option for XIAP deficiency, have improved. The pathophysiology of XIAP deficiency is not fully understood, however it is known that XIAP plays a role in both the innate and adaptive immune response and in immune regulation, most notably through modulation of tumour necrosis factor (TNF)-receptor signalling and regulation of NLRP3 inflammasome activity. In this review we will provide an up to date overview of both the clinical aspects and pathophysiology of XIAP deficiency.
Keywords: BIRC4; NOD2; X-linked lymphoproliferative disease; XIAP deficiency; haematopoietic stem cell transplantation; haemophagocytic lymphohistiocytosis; inflammasome; inflammatory bowel disease.
Copyright © 2021 Mudde, Booth and Marsh.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. (2010) 116:1079–82. 10.1182/blood-2010-01-256099 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources