

Computational study on the reactivity of imidazolium-functionalized manganese bipyridyl tricarbonyl electrocatalysts [Mn[bpyMe(Im-R)](CO)3Br]+ (R = Me, Me2 and Me4) for CO2-to-CO conversion over H2 formation
- PMID: 34223846
- PMCID: PMC10229143
- DOI: 10.1039/d1cp01576a
Computational study on the reactivity of imidazolium-functionalized manganese bipyridyl tricarbonyl electrocatalysts [Mn[bpyMe(Im-R)](CO)3Br]+ (R = Me, Me2 and Me4) for CO2-to-CO conversion over H2 formation
Abstract
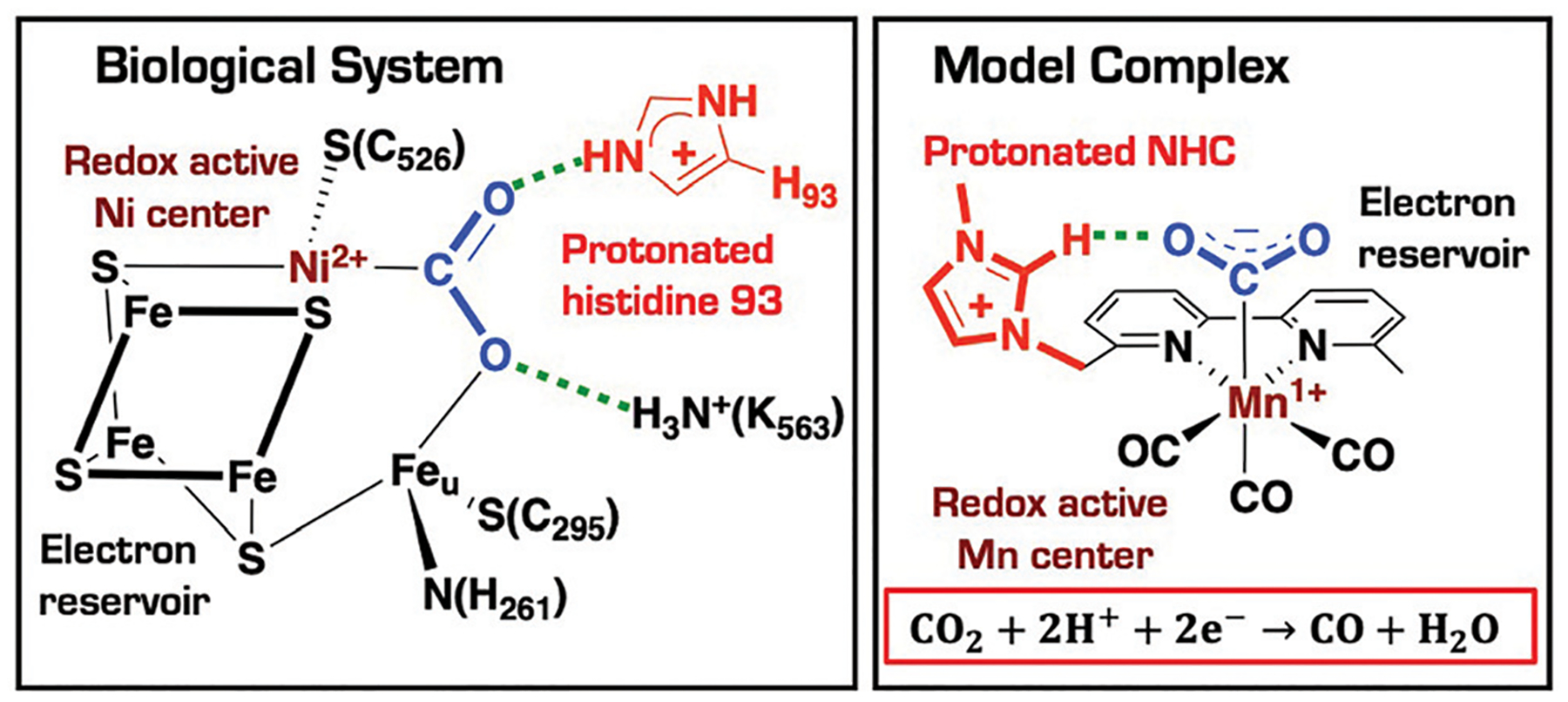
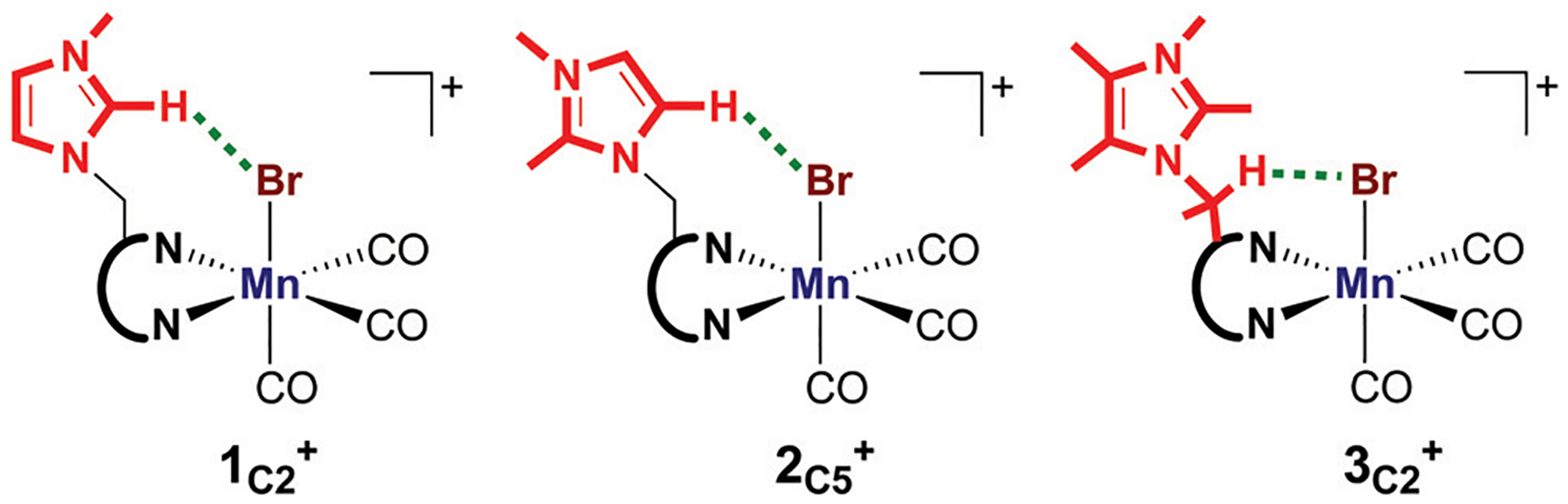
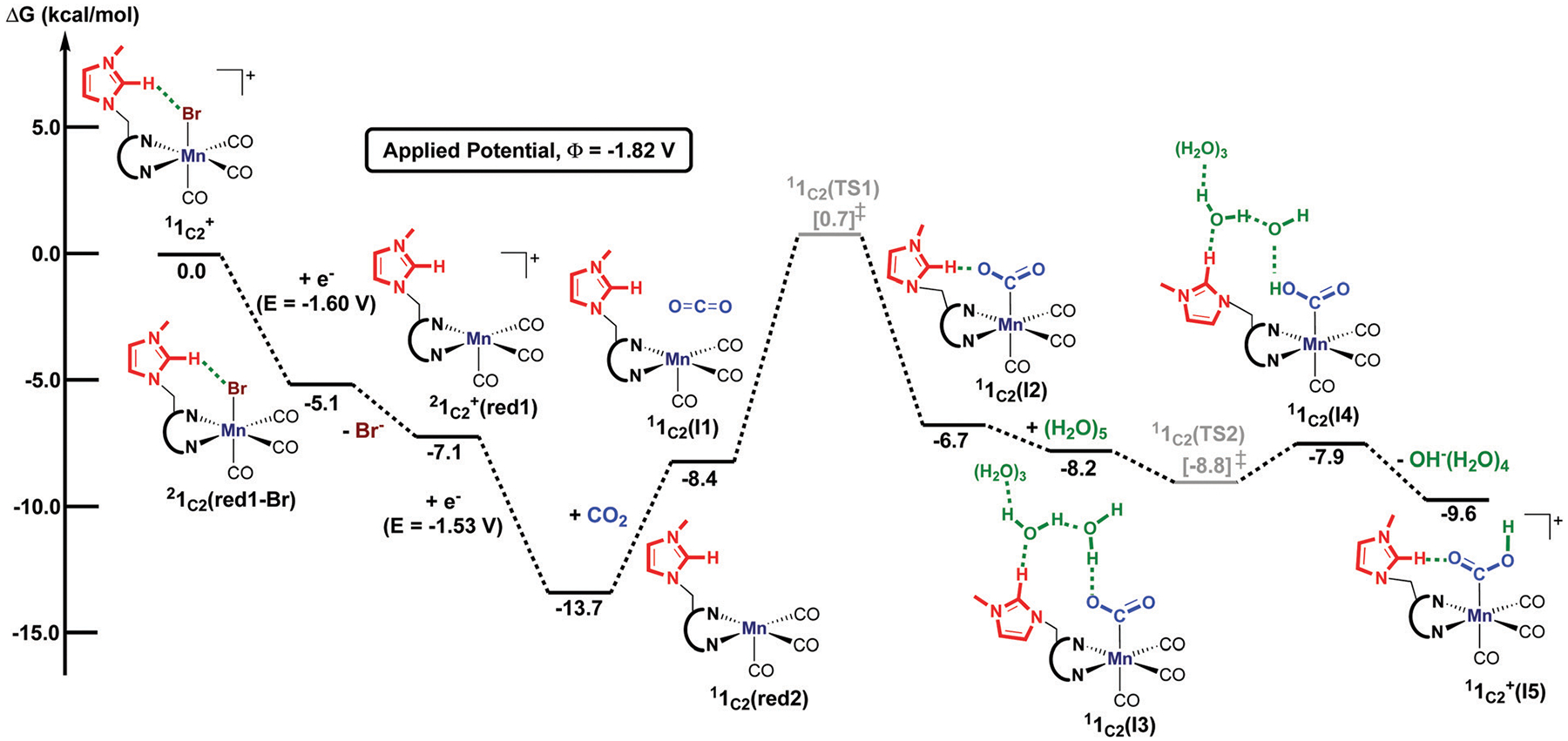
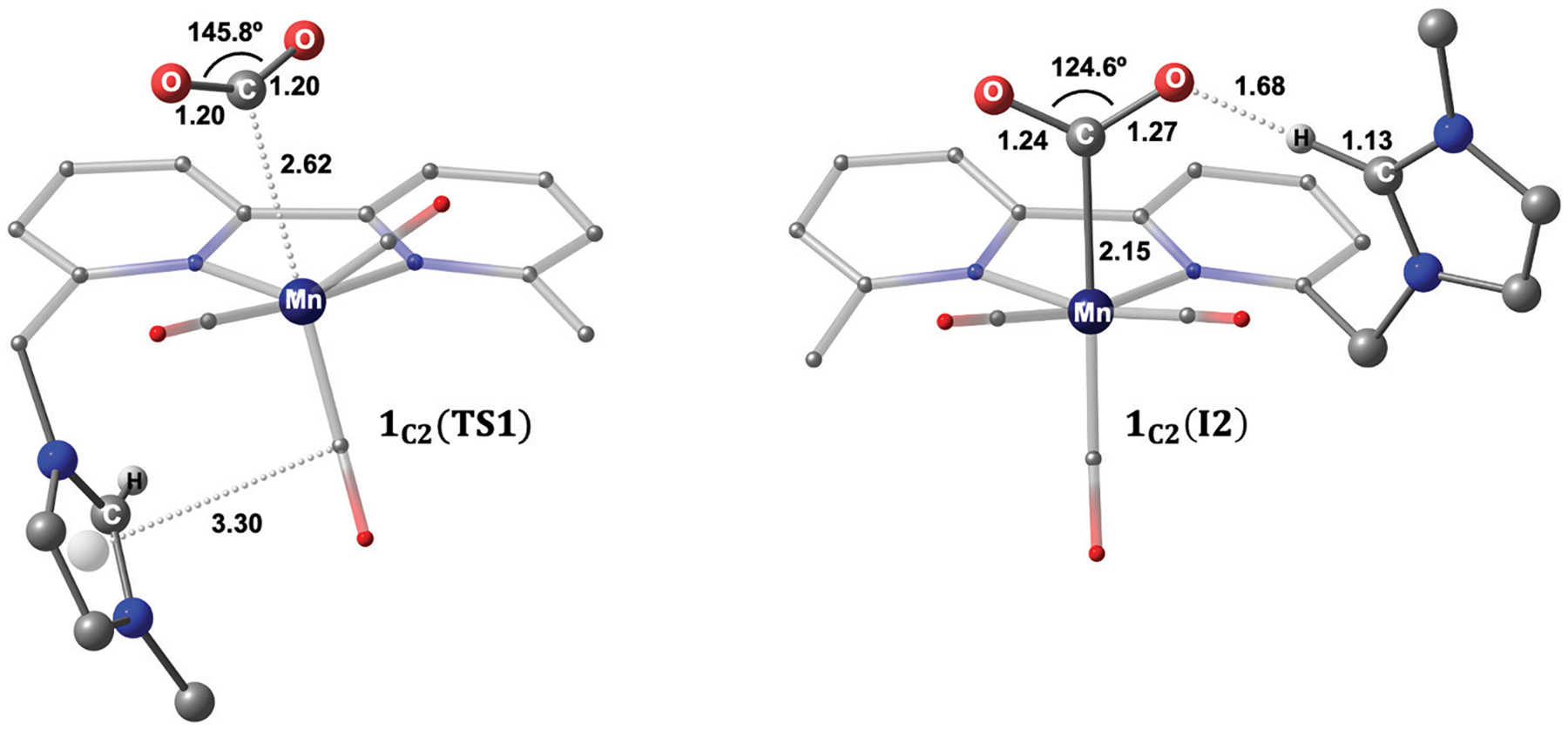
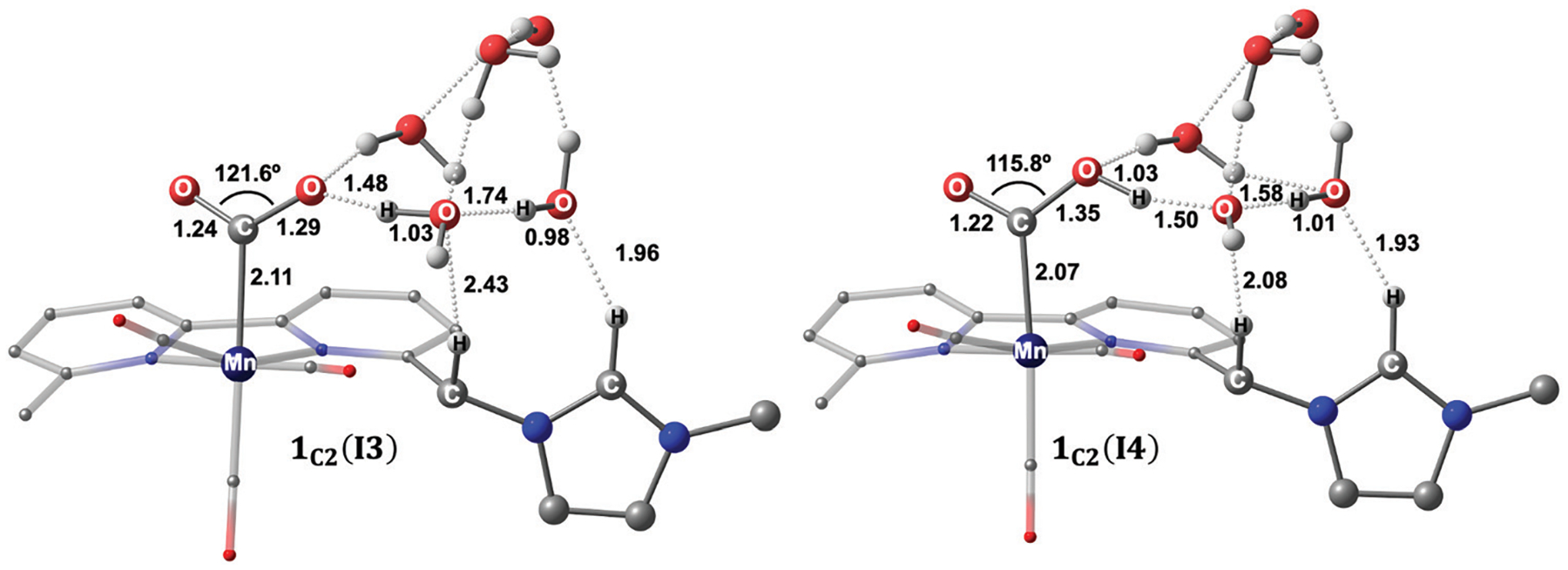
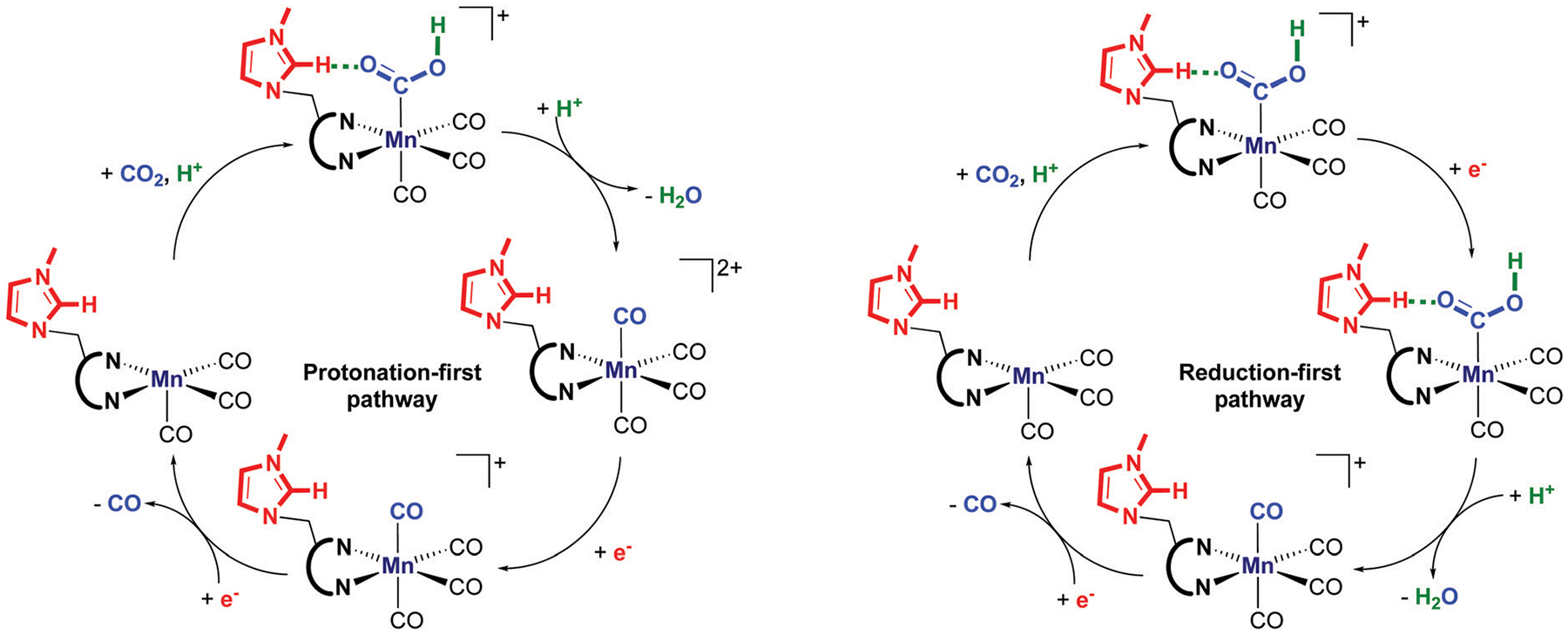
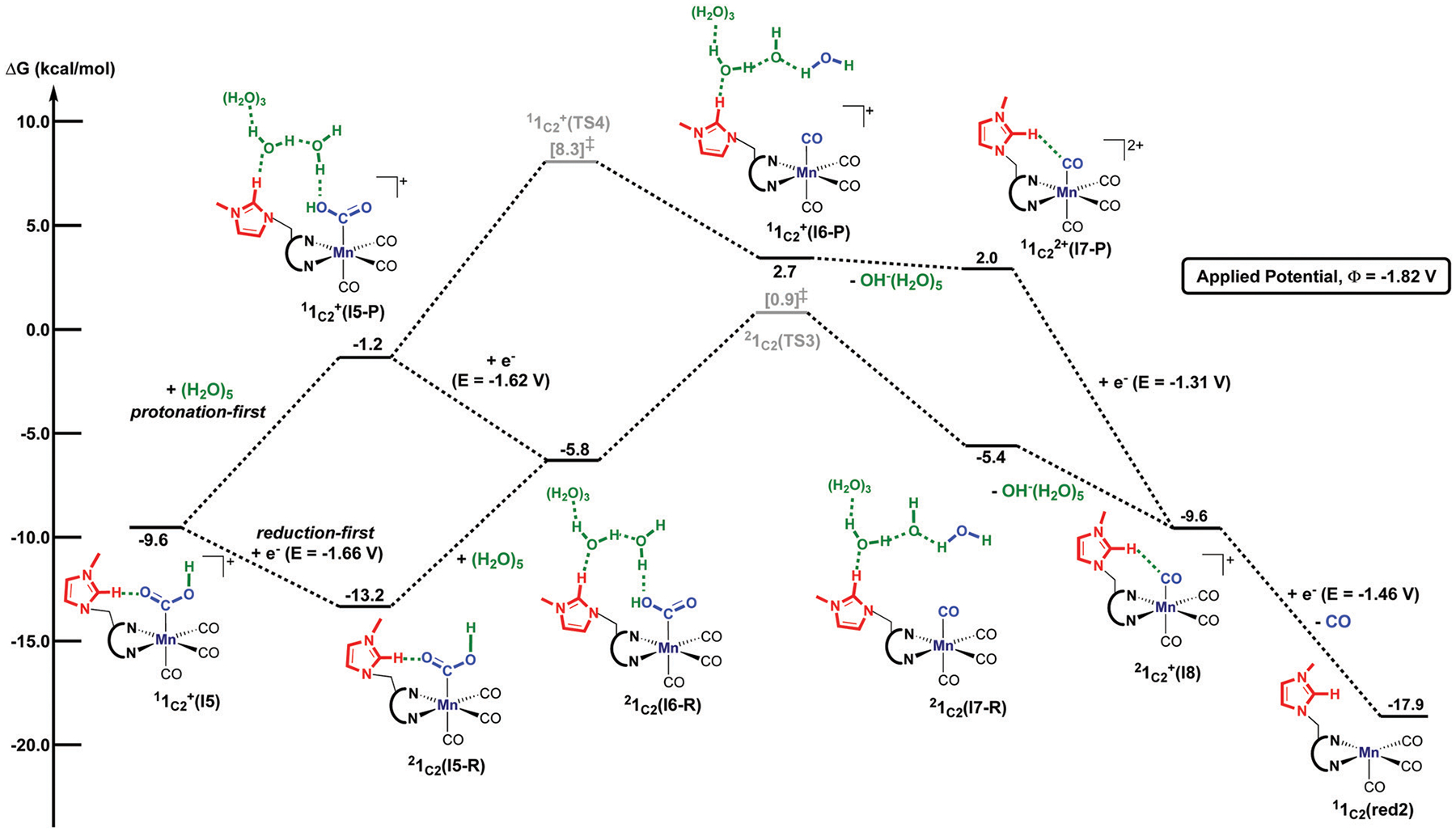
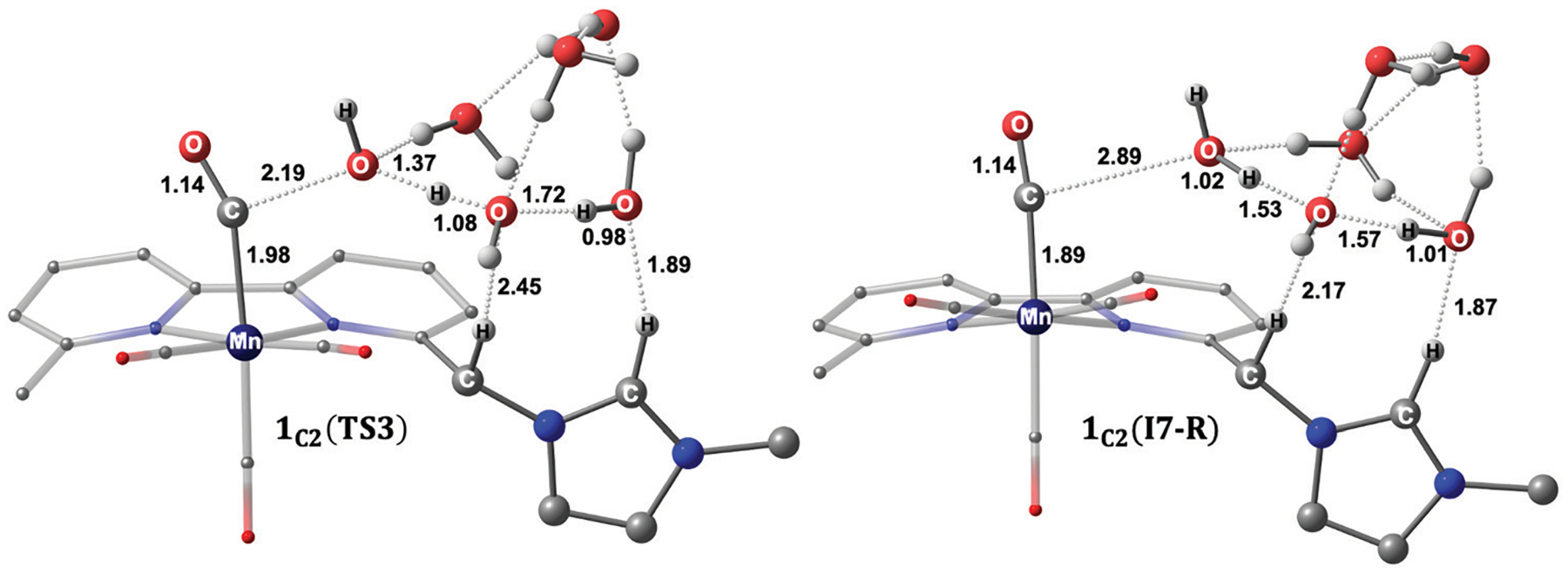
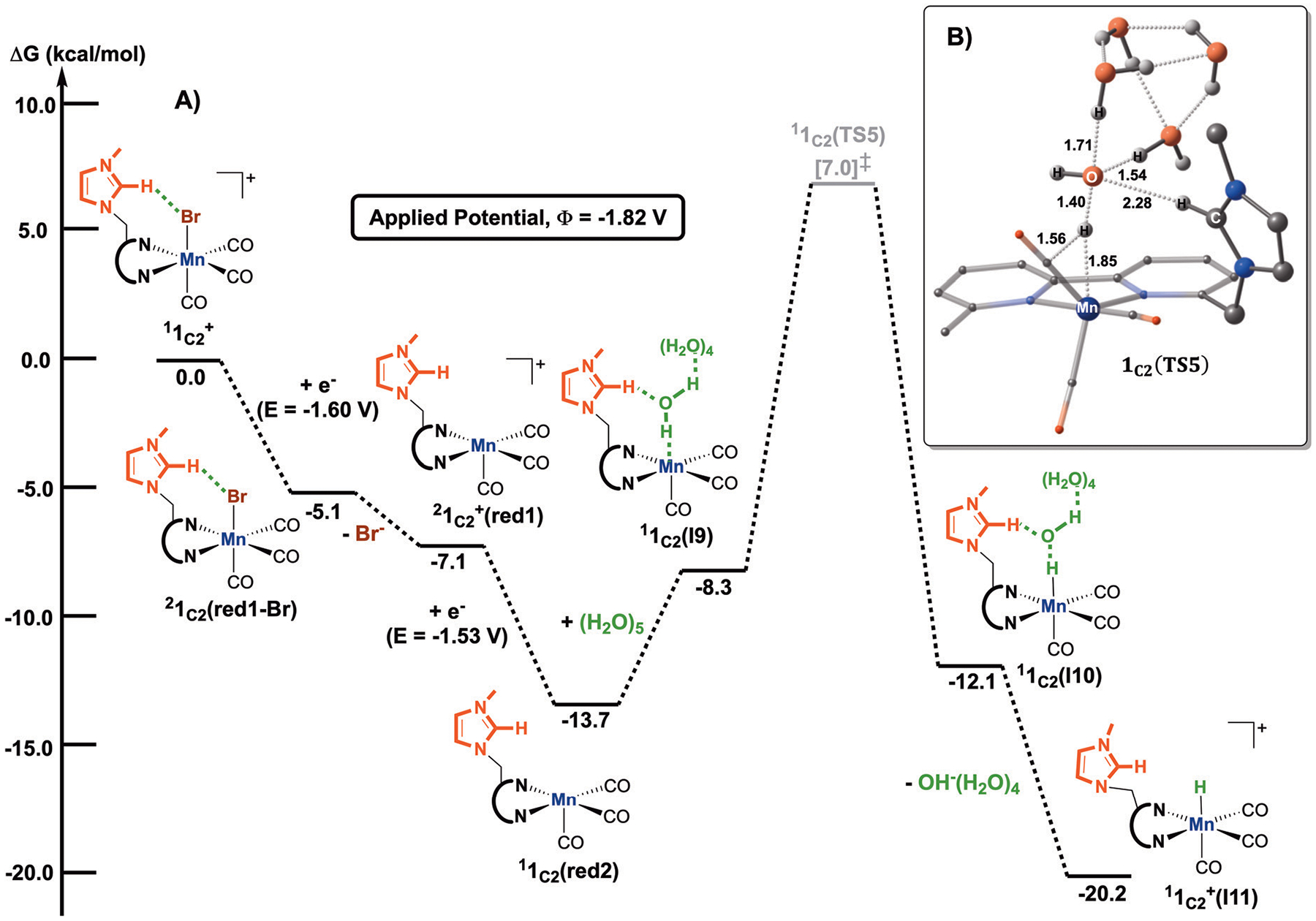
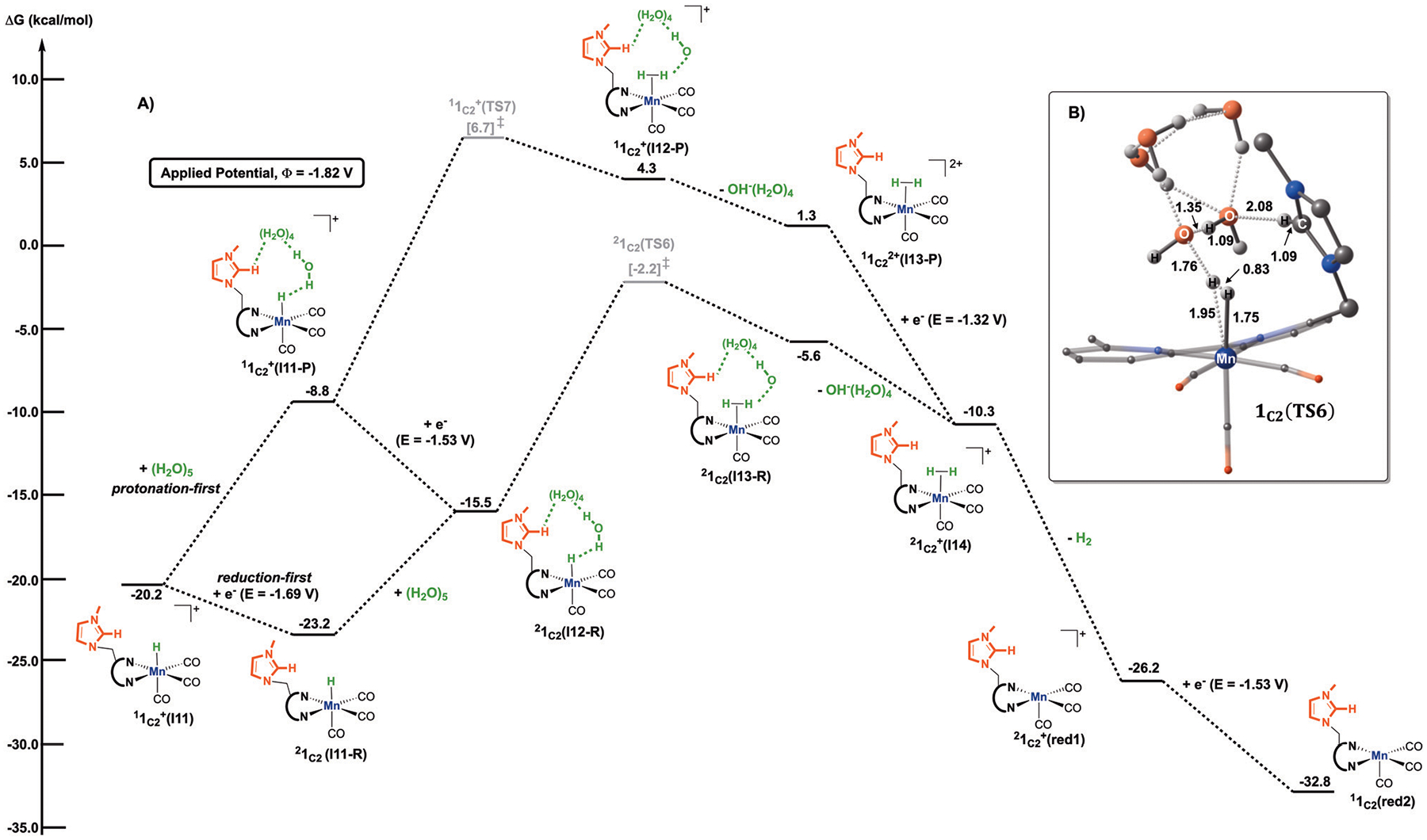
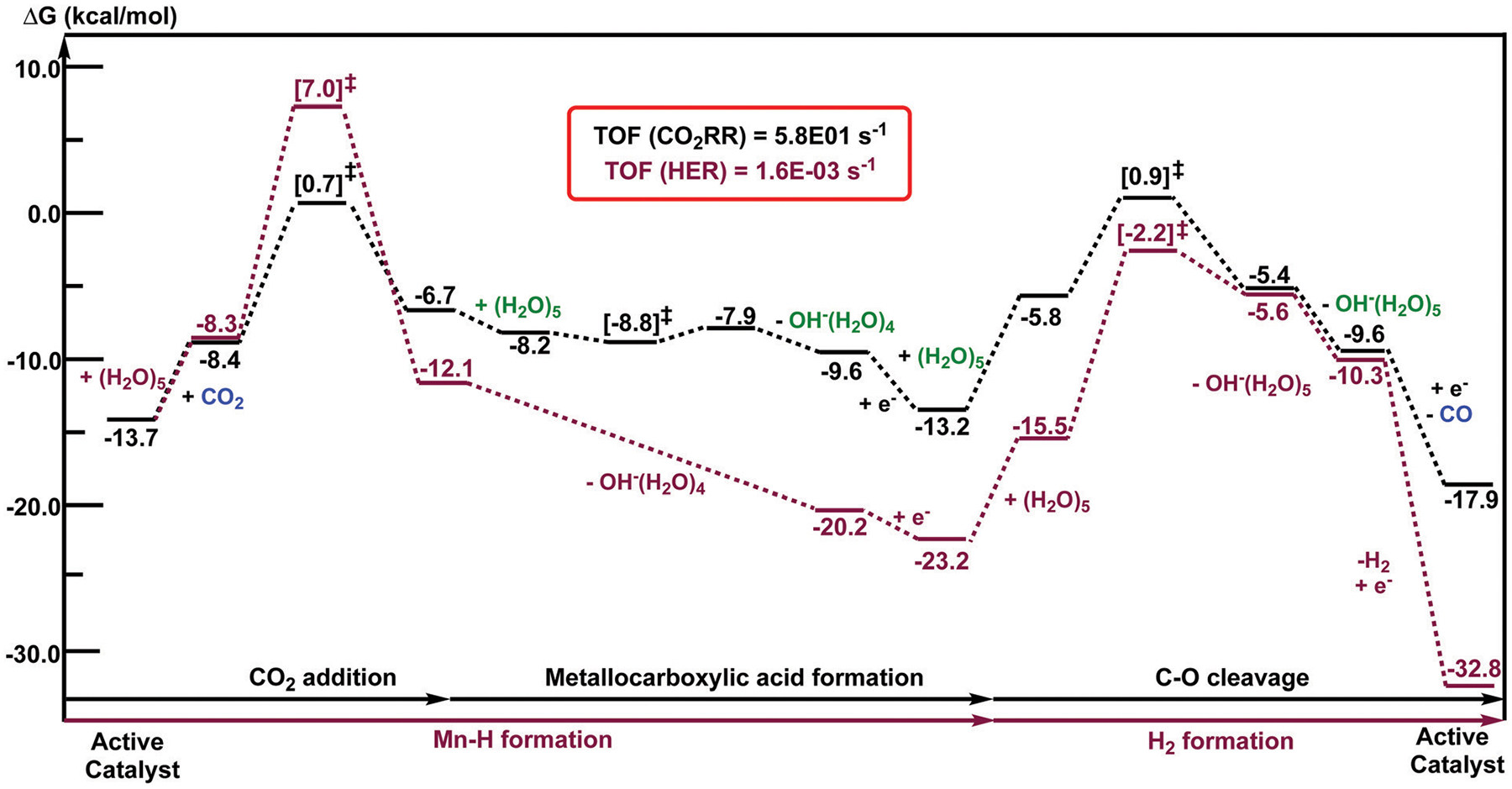
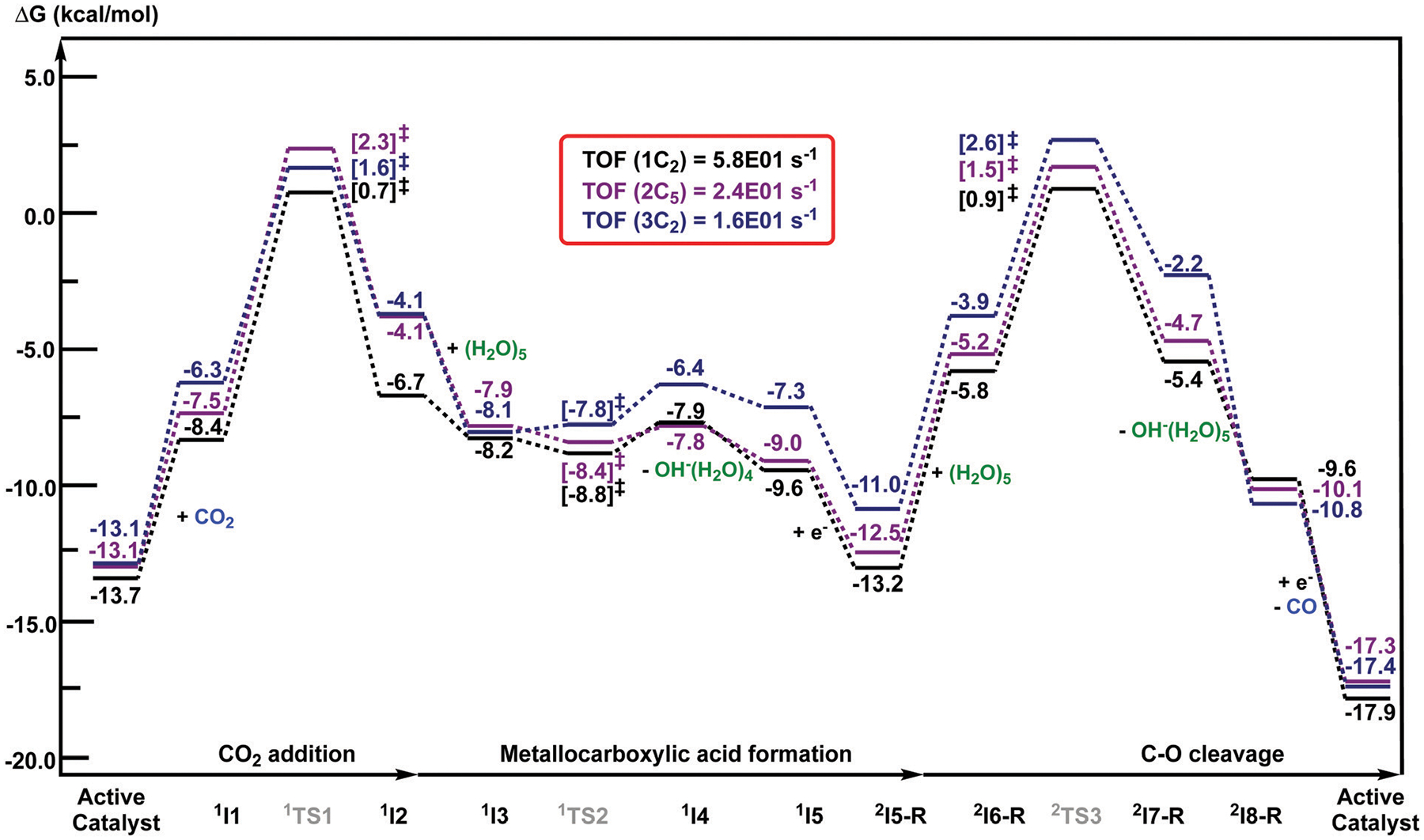
We have recently reported a series of imidazolium-functionalized manganese bipyridyl tricarbonyl electrocatalysts, [Mn[bpyMe(Im-R)](CO)3Br]+ (R = Me, Me2, and Me4), for CO2-to-CO conversion in the presence of H2O as the proton source [J. Am. Chem. Soc., 2019, 141, 6569]. These catalysts feature slightly acidic imidazolium moieties in the secondary coordination sphere and reduce CO2 at mild electrochemical potentials. Here, we employ density functional theory (DFT) calculations to understand the electronic structure and reactivity for the CO2 reduction reaction (CO2RR) over the competing hydrogen evolution reaction (HER) using [Mn[bpyMe(ImMe)](CO)3Br]+ (1+). Our work indicates that, in the absence of water, the imidazolium ligand stabilizes the Mn-CO2 adduct through hydrogen bonding-like interactions, similar to the activated CO2 molecule in the C-cluster of the Ni,Fe-carbon monoxide dehydrogenase II, and assists the protonation steps during CO2RR and HER. More significantly, based on the energy span model, we demonstrate that the selectivity for CO2 fixation over proton reduction results from a higher activation energy for yielding the manganese dihydrogen intermediate before H2 release, which is the TOF determining transition state (TDTS) under an applied potential of Φ = -1.82 V versus Fc0/+. The calculated TOF also reflects the selectivity for CO2RR, which is four orders of magnitude larger than for HER, consistent with the CPE experiments that show no hydrogen was obtained. In the case of CO2 reduction, the TOF determining intermediate (TDI) corresponds to the doubly reduced active catalyst, 1C2(red2), which features a manganese(0) center that couples ferromagnetically with one unpaired electron in the π* orbital of bipyridine. On the other hand, for HER, the metal-hydride intermediate, 1C2(I11-R), is the TDI. Finally, second-order perturbation analyses imply that the strongest hydrogen bonding-like interaction at the C2 position in 1+ contributes to the higher catalytic activity with respect to [Mn[bpyMe(ImMe2)](CO)3Br]+ (2+) and [Mn[bpyMe(ImMe4)](CO)3Br]+ (3+) for CO2 fixation, consistent with the experimental data.
Conflict of interest statement
Conflicts of interest
The authors declare no competing financial interest, and there are no conflicts of interest to declare.
Figures
Similar articles
-
Mechanistic Study of Tungsten Bipyridyl Tetracarbonyl Electrocatalysts for CO2 Fixation: Exploring the Roles of Explicit Proton Sources and Substituent Effects.Top Catal. 2022 Feb;65(1-4):325-340. doi: 10.1007/s11244-021-01529-7. Epub 2021 Nov 16. Top Catal. 2022. PMID: 37645456 Free PMC article.
-
Computational Study for CO2-to-CO Conversion over Proton Reduction Using [Re[bpyMe(Im-R)](CO)3Cl]+ (R = Me, Me2, and Me4) Electrocatalysts and Comparison with Manganese Analogues.ACS Catal. 2021 Nov 5;11(21):12989-13000. Epub 2021 Oct 12. ACS Catal. 2021. PMID: 36860803 Free PMC article.
-
Understanding the Role of Inter- and Intramolecular Promoters in Electro- and Photochemical CO2 Reduction Using Mn, Re, and Ru Catalysts.Acc Chem Res. 2022 Mar 1;55(5):616-628. doi: 10.1021/acs.accounts.1c00616. Epub 2022 Feb 8. Acc Chem Res. 2022. PMID: 35133133
-
Computational Modeling of Electrocatalysts for CO2 Reduction: Probing the Role of Primary, Secondary, and Outer Coordination Spheres.Acc Chem Res. 2025 Feb 4;58(3):342-353. doi: 10.1021/acs.accounts.4c00631. Epub 2025 Jan 27. Acc Chem Res. 2025. PMID: 39869093
-
Main group elements in electrochemical hydrogen evolution and carbon dioxide reduction.Chem Commun (Camb). 2023 Oct 4;59(79):11767-11779. doi: 10.1039/d3cc03606e. Chem Commun (Camb). 2023. PMID: 37695110 Review.
Cited by
-
Mechanistic Study of Tungsten Bipyridyl Tetracarbonyl Electrocatalysts for CO2 Fixation: Exploring the Roles of Explicit Proton Sources and Substituent Effects.Top Catal. 2022 Feb;65(1-4):325-340. doi: 10.1007/s11244-021-01529-7. Epub 2021 Nov 16. Top Catal. 2022. PMID: 37645456 Free PMC article.
-
Computational Study for CO2-to-CO Conversion over Proton Reduction Using [Re[bpyMe(Im-R)](CO)3Cl]+ (R = Me, Me2, and Me4) Electrocatalysts and Comparison with Manganese Analogues.ACS Catal. 2021 Nov 5;11(21):12989-13000. Epub 2021 Oct 12. ACS Catal. 2021. PMID: 36860803 Free PMC article.
-
Why surface hydrophobicity promotes CO2 electroreduction: a case study of hydrophobic polymer N-heterocyclic carbenes.Chem Sci. 2023 Aug 8;14(36):9664-9677. doi: 10.1039/d3sc02658b. eCollection 2023 Sep 20. Chem Sci. 2023. PMID: 37736633 Free PMC article.
-
Metal-Dependent Mechanism of the Electrocatalytic Reduction of CO2 by Bipyridine Complexes Bearing Pendant Amines: A DFT Study.ACS Org Inorg Au. 2024 Oct 11;5(1):26-36. doi: 10.1021/acsorginorgau.4c00046. eCollection 2025 Feb 5. ACS Org Inorg Au. 2024. PMID: 39927101 Free PMC article.
References
-
- Chu S, Cui Y and Liu N, Nat. Mater, 2016, 16, 16–22. - PubMed
-
- Feely RA, Sabine CL, Lee K, Berelson W, Kleypas J, Fabry VJ and Millero FJ, Science, 2004, 305, 362–366. - PubMed
-
- Berry HL, Bowen K and Kjellstrom T, Int. J. Public Health, 2010, 55, 123–132. - PubMed
-
- Bourque F and Cunsolo Willox A, Int. Rev. Psychiatry, 2014, 26, 415–422. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous