

Mako: A Graph-based Pattern Growth Approach to Detect Complex Structural Variants
- PMID: 34224879
- PMCID: PMC9510932
- DOI: 10.1016/j.gpb.2021.03.007
Mako: A Graph-based Pattern Growth Approach to Detect Complex Structural Variants
Abstract
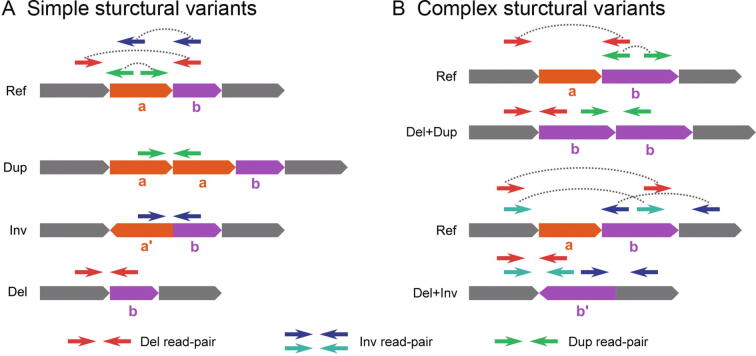
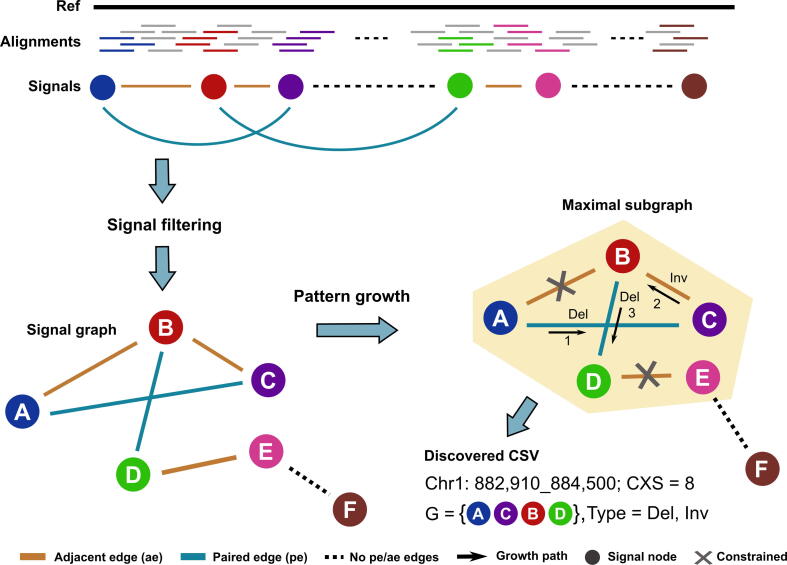
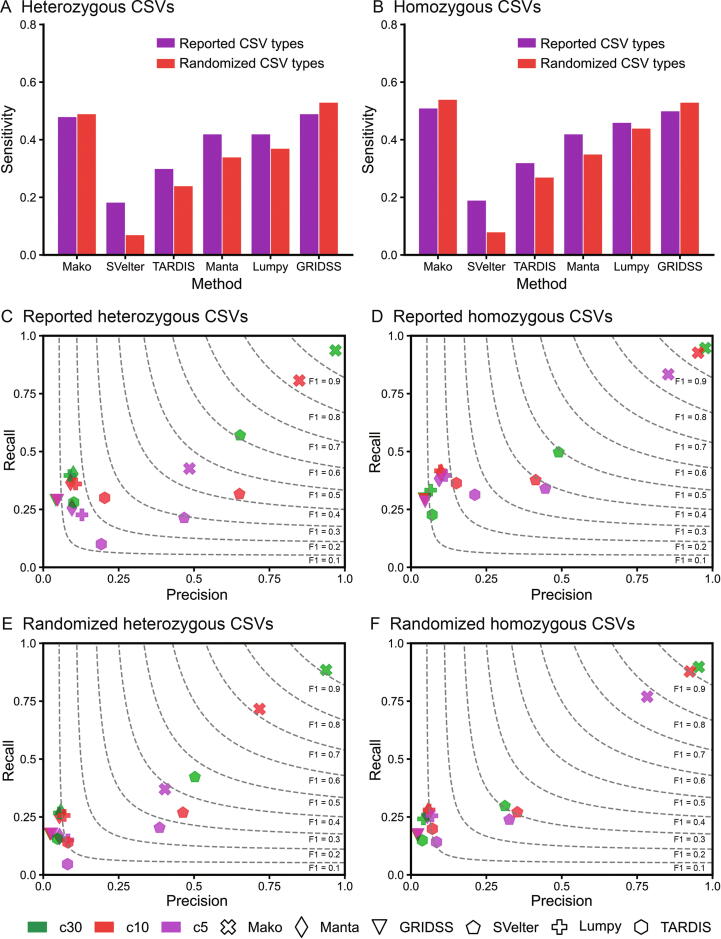
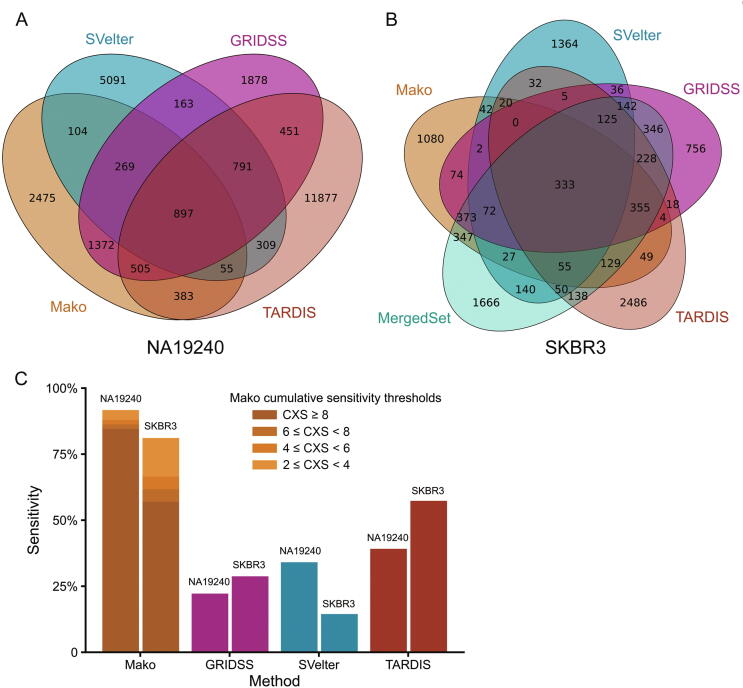
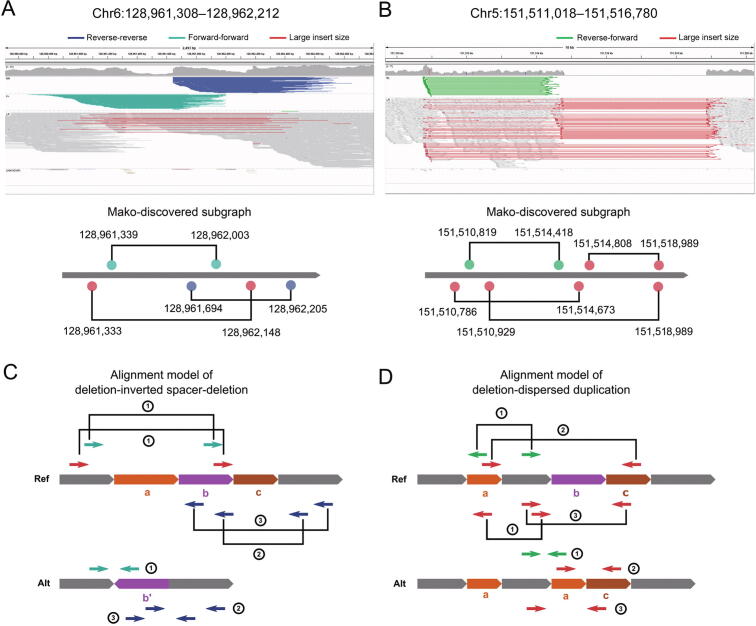
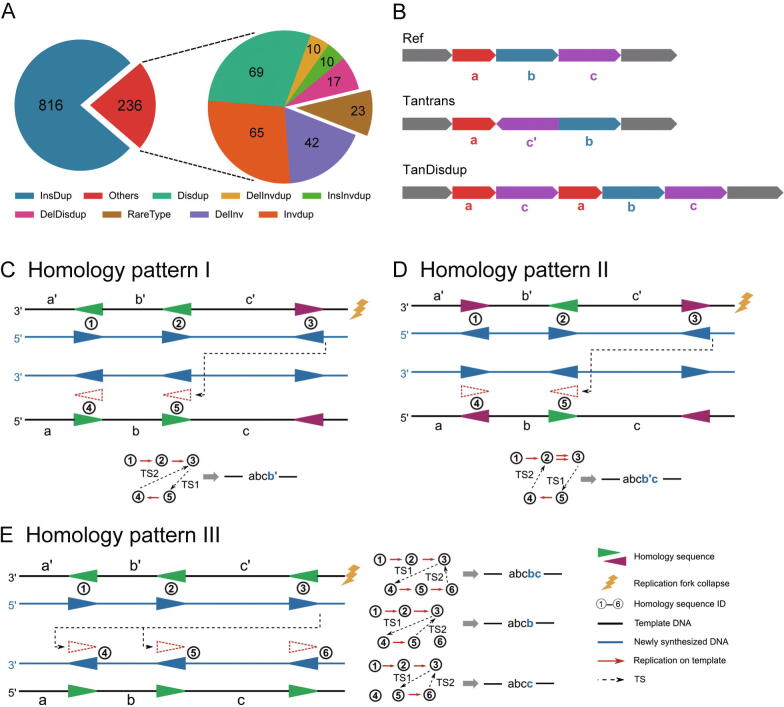
Complex structural variants (CSVs) are genomic alterations that have more than two breakpoints and are considered as the simultaneous occurrence of simple structural variants. However, detecting the compounded mutational signals of CSVs is challenging through a commonly used model-match strategy. As a result, there has been limited progress for CSV discovery compared with simple structural variants. Here, we systematically analyzed the multi-breakpoint connection feature of CSVs, and proposed Mako, utilizing a bottom-up guided model-free strategy, to detect CSVs from paired-end short-read sequencing. Specifically, we implemented a graph-based pattern growth approach, where the graph depicts potential breakpoint connections, and pattern growth enables CSV detection without pre-defined models. Comprehensive evaluations on both simulated and real datasets revealed that Mako outperformed other algorithms. Notably, validation rates of CSVs on real data based on experimental and computational validations as well as manual inspections are around 70%, where the medians of experimental and computational breakpoint shift are 13 bp and 26 bp, respectively. Moreover, the Mako CSV subgraph effectively characterized the breakpoint connections of a CSV event and uncovered a total of 15 CSV types, including two novel types of adjacent segment swap and tandem dispersed duplication. Further analysis of these CSVs also revealed the impact of sequence homology on the formation of CSVs. Mako is publicly available at https://github.com/xjtu-omics/Mako.
Keywords: Complex structural variant; Formation mechanism; Graph mining; Next-generation sequencing; Pattern growth.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Competing Interests The authors have declared no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
