

Coordinated changes in gene expression kinetics underlie both mouse and human erythroid maturation
- PMID: 34225769
- PMCID: PMC8258993
- DOI: 10.1186/s13059-021-02414-y
Coordinated changes in gene expression kinetics underlie both mouse and human erythroid maturation
Abstract
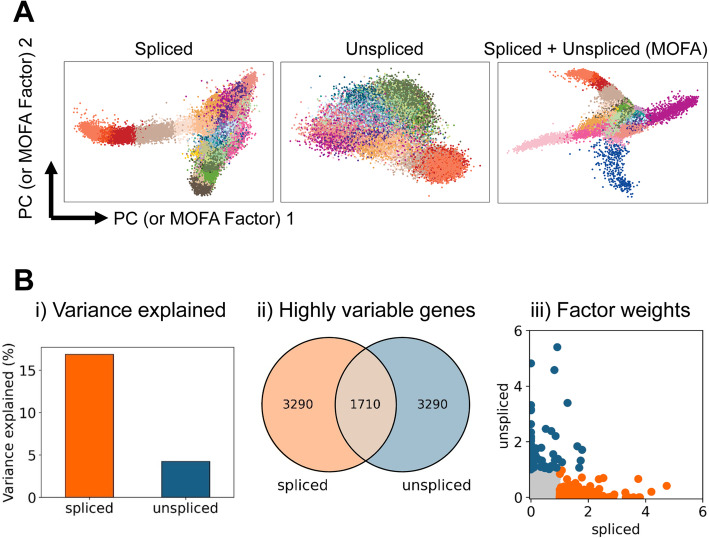
Background: Single-cell technologies are transforming biomedical research, including the recent demonstration that unspliced pre-mRNA present in single-cell RNA-Seq permits prediction of future expression states. Here we apply this RNA velocity concept to an extended timecourse dataset covering mouse gastrulation and early organogenesis.
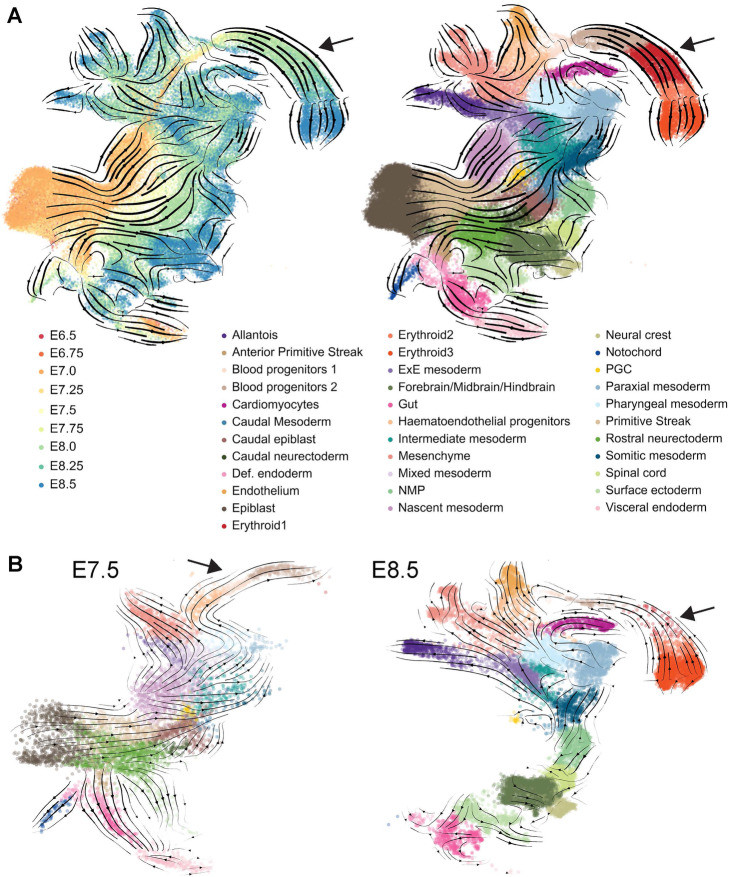
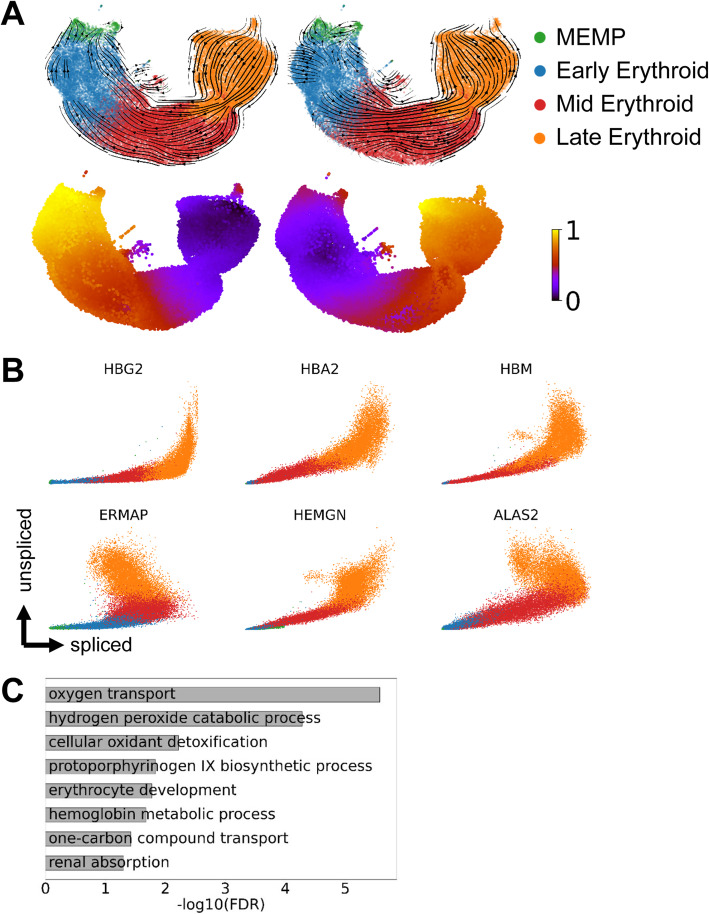
Results: Intriguingly, RNA velocity correctly identifies epiblast cells as the starting point, but several trajectory predictions at later stages are inconsistent with both real-time ordering and existing knowledge. The most striking discrepancy concerns red blood cell maturation, with velocity-inferred trajectories opposing the true differentiation path. Investigating the underlying causes reveals a group of genes with a coordinated step-change in transcription, thus violating the assumptions behind current velocity analysis suites, which do not accommodate time-dependent changes in expression dynamics. Using scRNA-Seq analysis of chimeric mouse embryos lacking the major erythroid regulator Gata1, we show that genes with the step-changes in expression dynamics during erythroid differentiation fail to be upregulated in the mutant cells, thus underscoring the coordination of modulating transcription rate along a differentiation trajectory. In addition to the expected block in erythroid maturation, the Gata1-chimera dataset reveals induction of PU.1 and expansion of megakaryocyte progenitors. Finally, we show that erythropoiesis in human fetal liver is similarly characterized by a coordinated step-change in gene expression.
Conclusions: By identifying a limitation of the current velocity framework coupled with in vivo analysis of mutant cells, we reveal a coordinated step-change in gene expression kinetics during erythropoiesis, with likely implications for many other differentiation processes.
Keywords: Erythropoiesis; Gastrulation; Gata1; RNA velocity.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
