

Improving statistical power in severe malaria genetic association studies by augmenting phenotypic precision
- PMID: 34225842
- PMCID: PMC8315799
- DOI: 10.7554/eLife.69698
Improving statistical power in severe malaria genetic association studies by augmenting phenotypic precision
Abstract
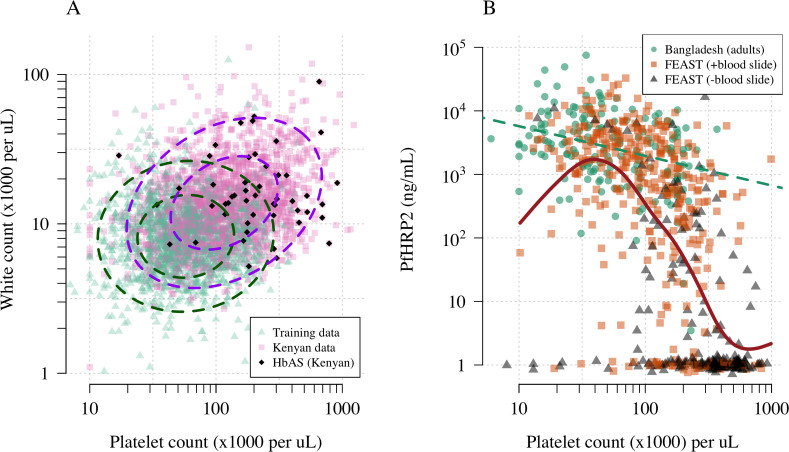
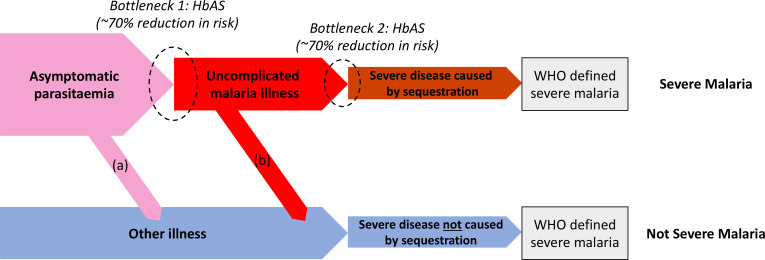
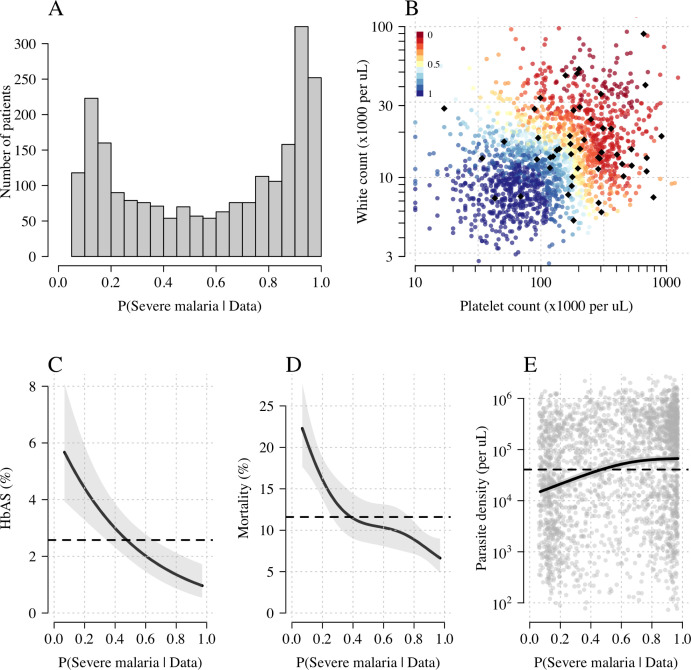
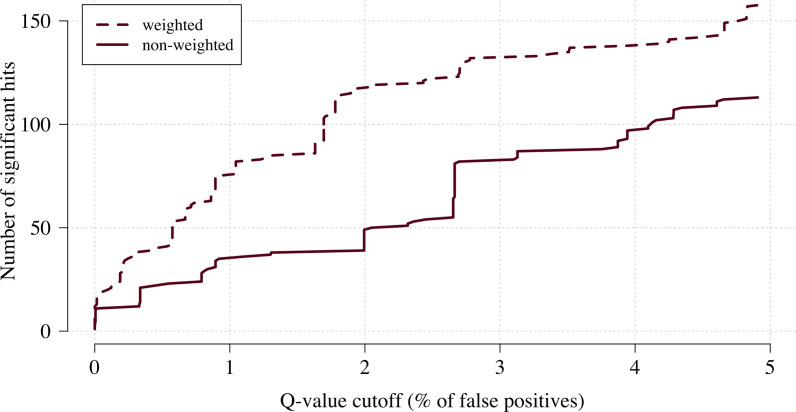
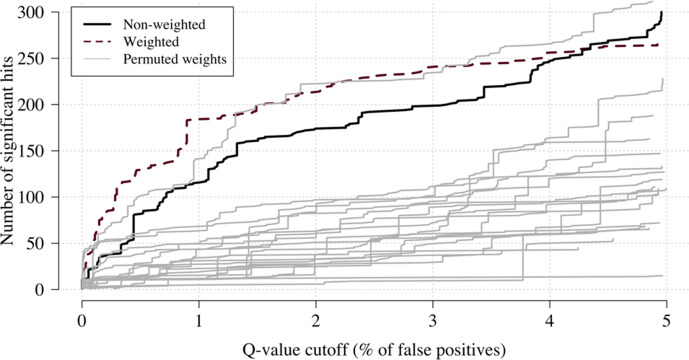
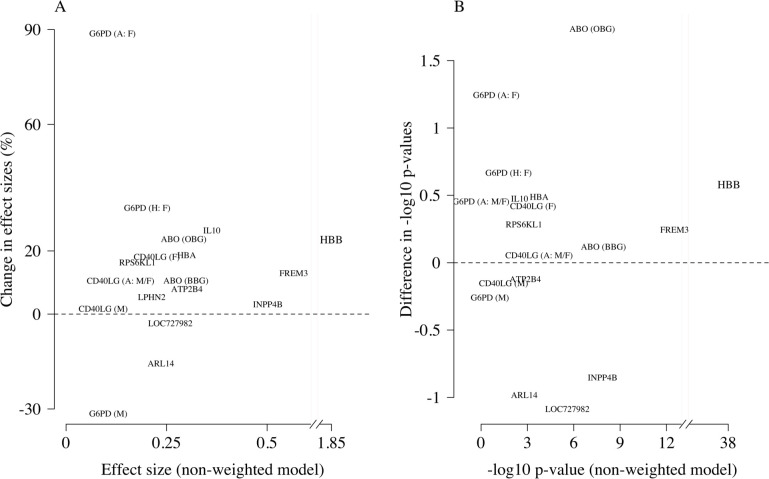
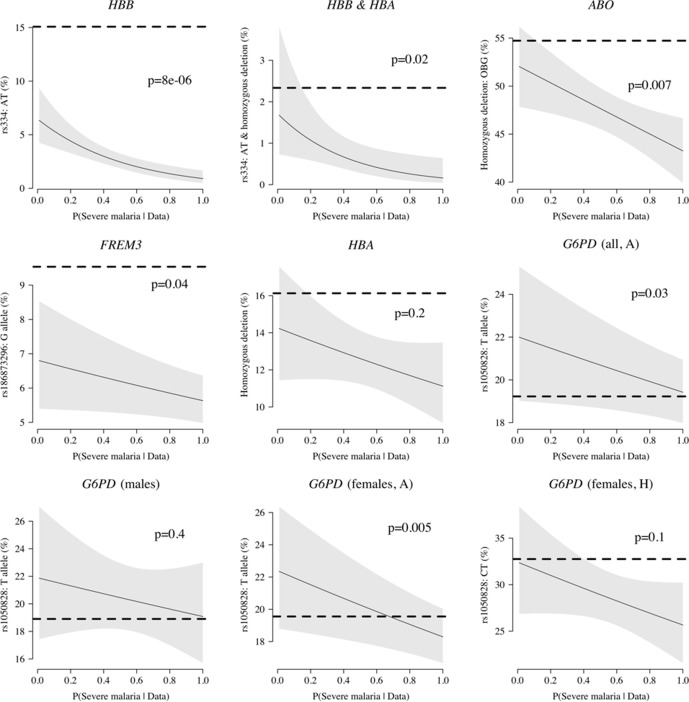
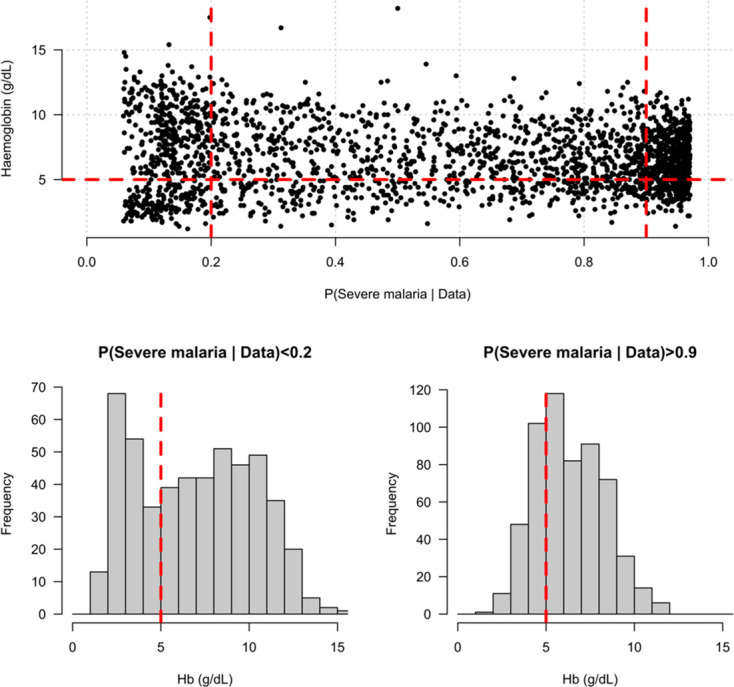
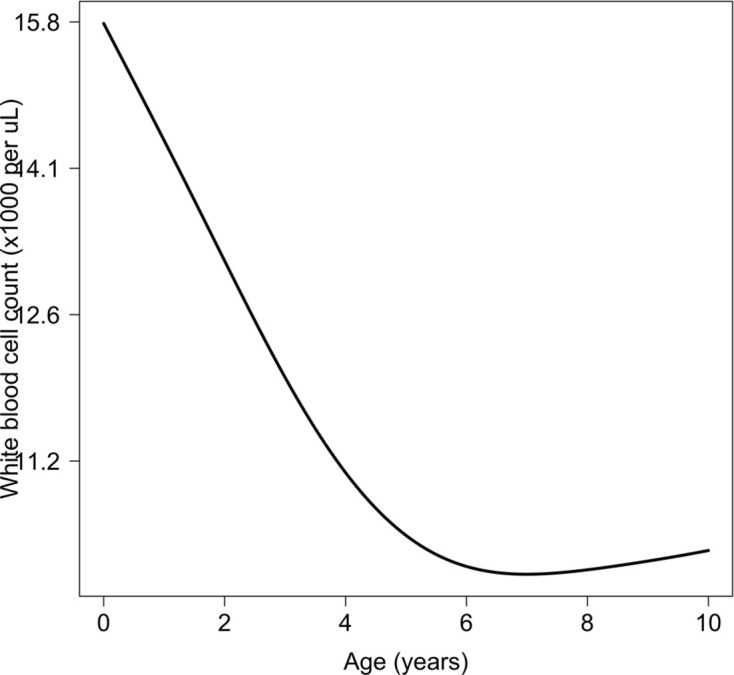
Severe falciparum malaria has substantially affected human evolution. Genetic association studies of patients with clinically defined severe malaria and matched population controls have helped characterise human genetic susceptibility to severe malaria, but phenotypic imprecision compromises discovered associations. In areas of high malaria transmission, the diagnosis of severe malaria in young children and, in particular, the distinction from bacterial sepsis are imprecise. We developed a probabilistic diagnostic model of severe malaria using platelet and white count data. Under this model, we re-analysed clinical and genetic data from 2220 Kenyan children with clinically defined severe malaria and 3940 population controls, adjusting for phenotype mis-labelling. Our model, validated by the distribution of sickle trait, estimated that approximately one-third of cases did not have severe malaria. We propose a data-tilting approach for case-control studies with phenotype mis-labelling and show that this reduces false discovery rates and improves statistical power in genome-wide association studies.
Keywords: GWAS; complete blood count; diagnosis; epidemiology; genetics; genomics; global health; human; severe malaria.
Plain language summary
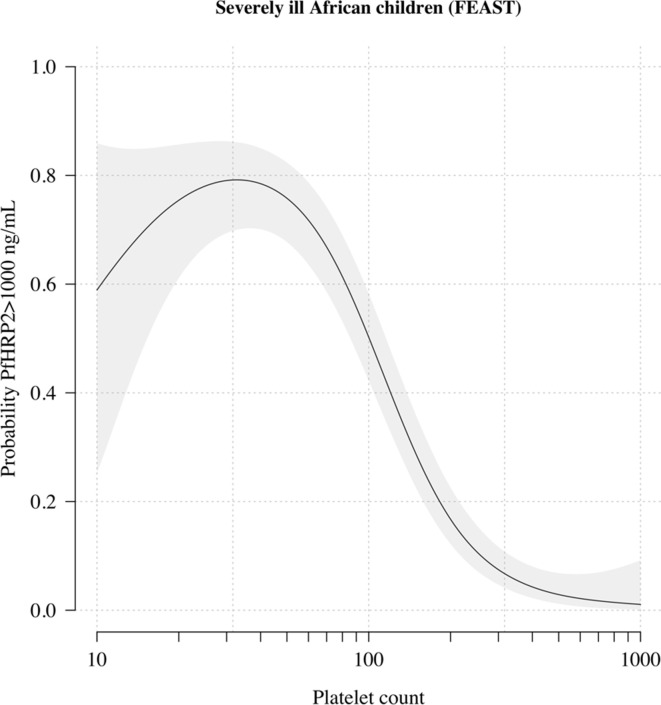
In areas of sub-Saharan Africa where malaria is common, most people are frequently exposed to the bites of mosquitoes carrying malaria parasites, so they often have malaria parasites in their blood. Young children, who have not yet built up strong immunity against malaria, often fall ill with severe malaria, a life-threatening disease. It is unclear why some children develop severe malaria and die, while other children with high numbers of parasites in their blood do not develop any apparent symptoms. Genetic susceptibility studies are designed to uncover why such differences exist by comparing individuals with severe malaria (referred to as ‘cases’) with individuals drawn from the general population (known as ‘controls’). But severe malaria can be a challenge to diagnose. Since high numbers of malaria parasites can be found in healthy children, it is sometimes difficult to determine whether the parasites are making a child ill, or whether they are a coincidental finding. Consequently, some of the ‘cases’ recruited into these studies may actually have a different disease, such as bacterial sepsis. This ultimately affects how the studies are interpreted, and introduces error and inaccuracy into the data. Watson, Ndila et al. investigated whether measuring blood biomarkers in patients (derived from the complete blood count, including platelet counts and white blood cell counts) could improve the accuracy with which malaria is diagnosed. They developed a new mathematical model that incorporates platelet and white blood cell counts. This model estimates that in a large cohort of 2,220 Kenyan children diagnosed with severe malaria, around one third of enrolled children did not actually have this disease. Further analysis suggests that patients with severe malaria are highly unlikely to have platelet counts higher than 200,000 per microlitre. This defines a cut-off that researchers can use to avoid recruiting patients who do not have severe malaria in future studies. Additionally, the ability to diagnose severe malaria more accurately can make it easier to detect and treat other diseases with similar symptoms in children with high numbers of malaria parasites in their blood. Watson, Ndila et al.’s findings support the recommendation that all children with suspected malaria be given broad spectrum antibiotics, as many misdiagnosed children will likely have bacterial sepsis. It also suggests that using complete blood counts, which are cheap to obtain and increasingly available in low-resource settings, could improve diagnostic accuracy in future clinical studies of severe malaria. This could ultimately improve the ability of these studies to find new treatments for this life-threatening disease.
© 2021, Watson et al.
Conflict of interest statement
JW, CN, SU, AM, GN, SM, CN, NM, NP, BT, KR, SL, HK, EG, KM, ND, AD, PB, TW, CH, NW No competing interests declared
Figures
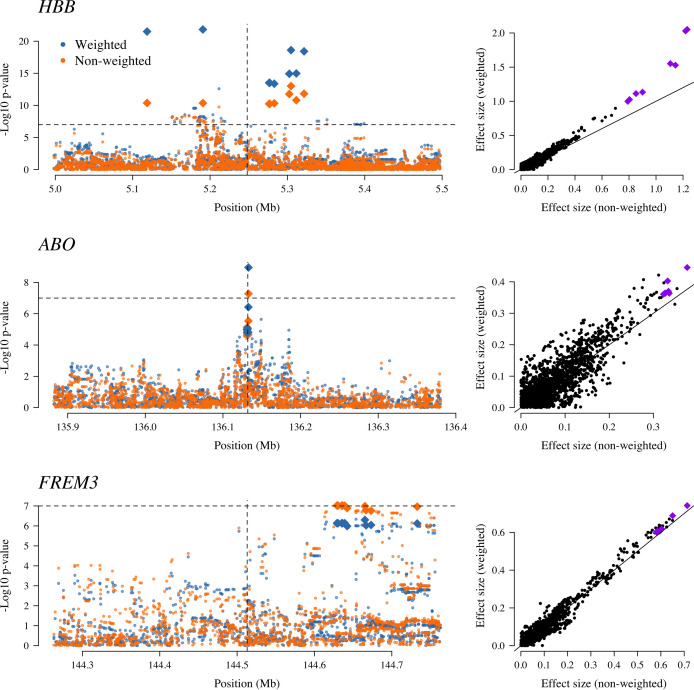
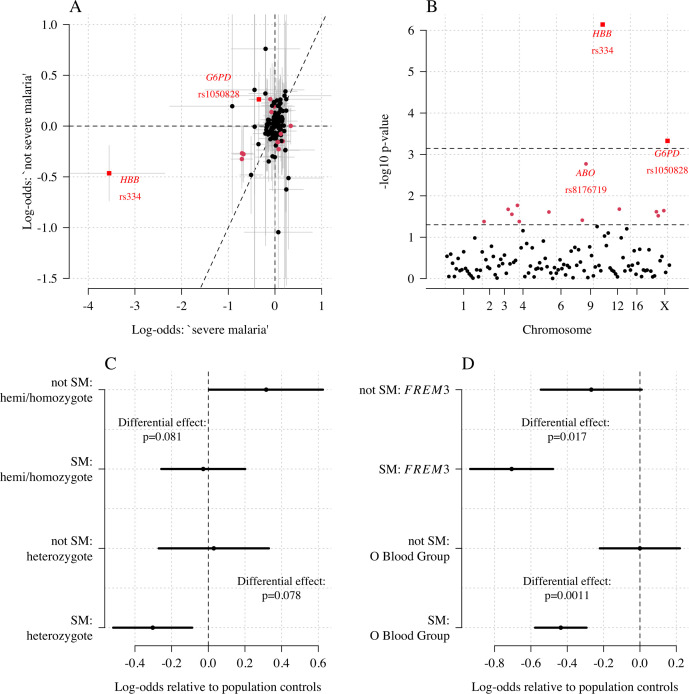
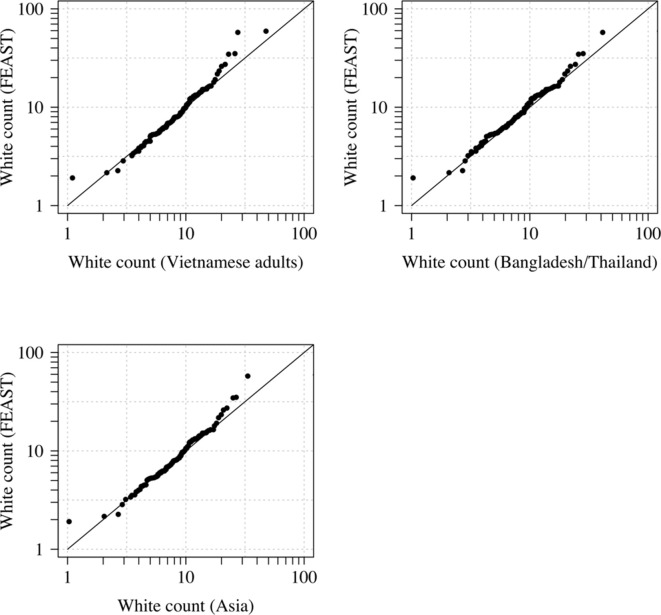
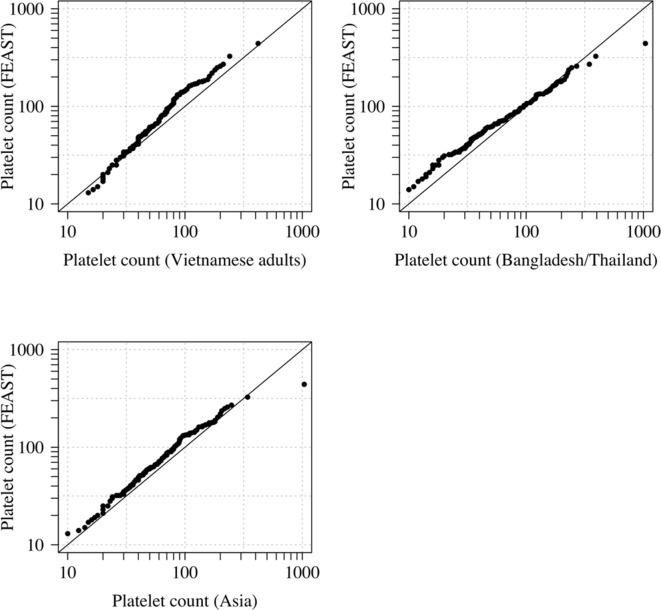
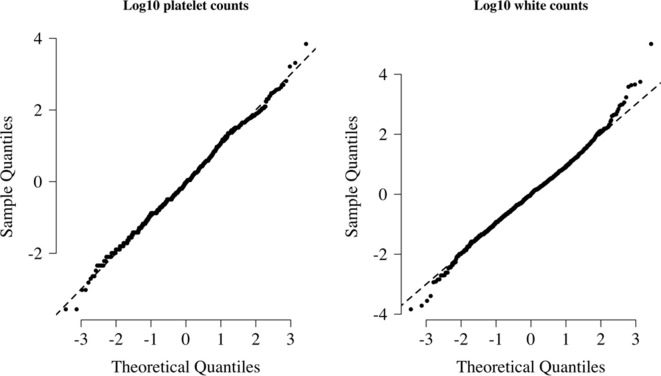
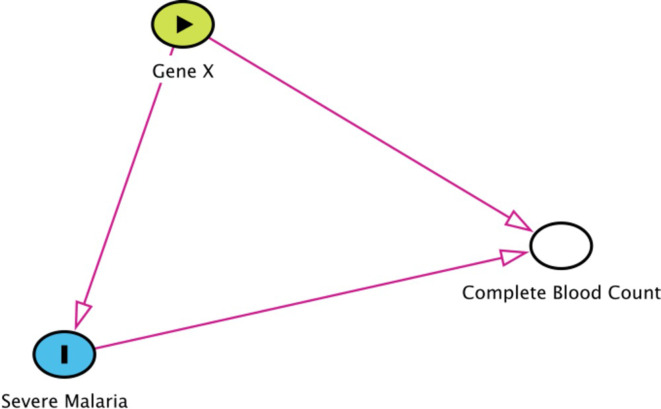
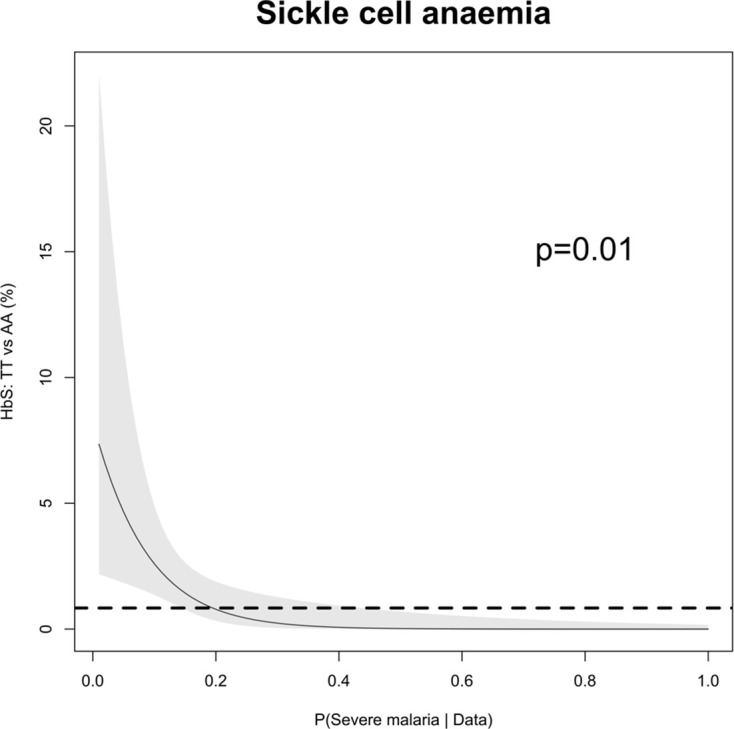
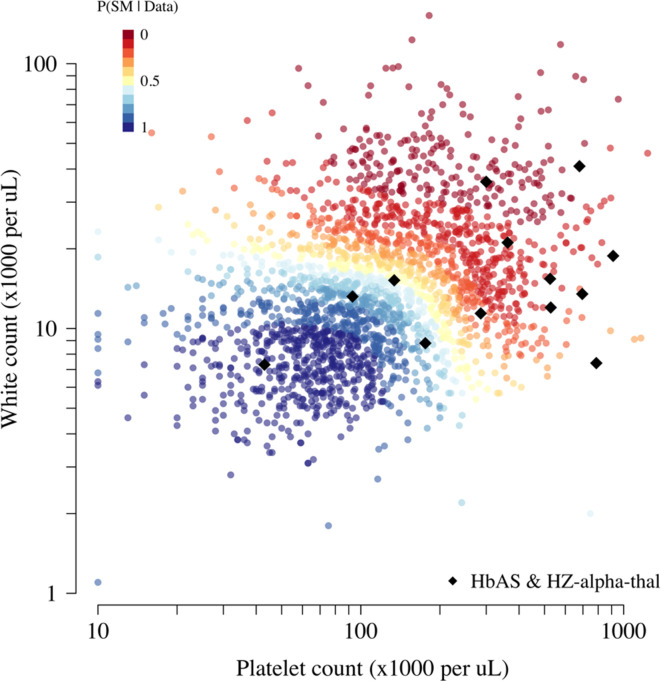
References
-
- Band G, Le QS, Clarke GM, Kivinen K, Hubbart C, Jeffreys AE, Rowlands K, Leffler EM, Jallow M, Conway DJ, Sisay-Joof F, Sirugo G, d’Alessandro U, Toure OB, Thera MA, Konate S, Sissoko S, Mangano VD, Bougouma EC, Sirima SB, Amenga-Etego LN, Ghansah AK, Hodgson AVO, Wilson MD, Enimil A, Ansong D, Evans J, Ademola SA, Apinjoh TO, Ndila CM, Manjurano A, Drakeley C, Reyburn H, Phu NH, Quyen NTN, Thai CQ, Hien TT, Teo YY, Manning L, Laman M, Michon P, Karunajeewa H, Siba P, Allen S, Allen A, Bahlo M, Davis TME, Simpson V, Shelton J, Spencer CCA, Busby GBJ, Kerasidou A, Drury E, Stalker J, Dilthey A, Mentzer AJ, McVean G, Bojang KA, Doumbo O, Modiano D, Koram KA, Agbenyega T, Amodu OK, Achidi E, Williams TN, Marsh K, Riley EM, Molyneux M, Taylor T, Dunstan SJ, Farrar J, Mueller I, Rockett KA, Kwiatkowski DP, Network MGE. Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania. Nature Communications. 2019;10:5732. doi: 10.1038/s41467-019-13480-z. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G0600230/MRC_/Medical Research Council/United Kingdom
- G0801439/MRC_/Medical Research Council/United Kingdom
- 206194/WT_/Wellcome Trust/United Kingdom
- WT077383/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- 204911/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- G0601027/MRC_/Medical Research Council/United Kingdom
- MC_UP_A390_1107/MRC_/Medical Research Council/United Kingdom
- MC_UU_00004/05/MRC_/Medical Research Council/United Kingdom
- 202800/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- G0600718/MRC_/Medical Research Council/United Kingdom
- MC_UU_12023/26/MRC_/Medical Research Council/United Kingdom
- 093956/Z/10/C/WT_/Wellcome Trust/United Kingdom
- 090770/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- MR/M006212/1/MRC_/Medical Research Council/United Kingdom
- 209265/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- 203141/Z/16/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
