

Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding
- PMID: 34226565
- PMCID: PMC8257795
- DOI: 10.1038/s41467-021-24328-w
Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding
Abstract
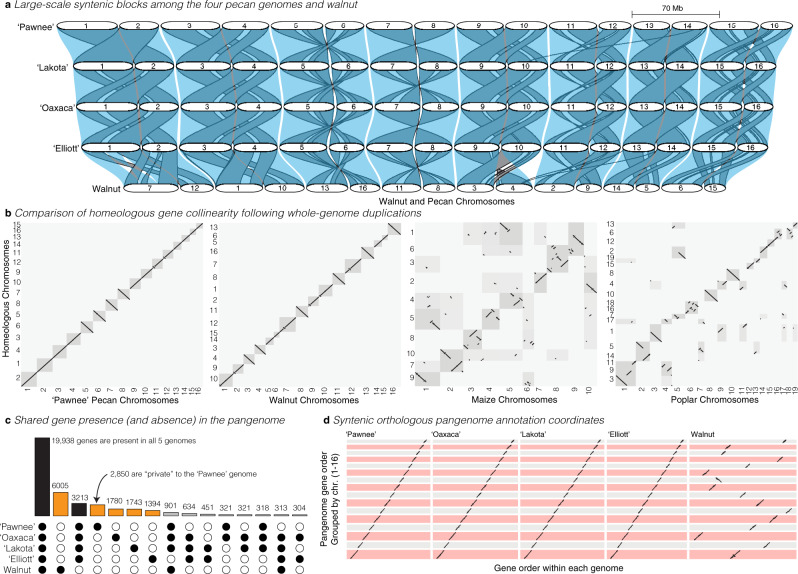
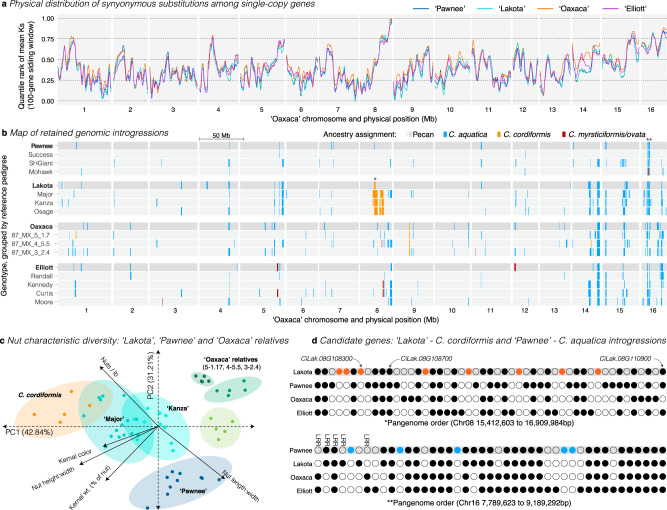
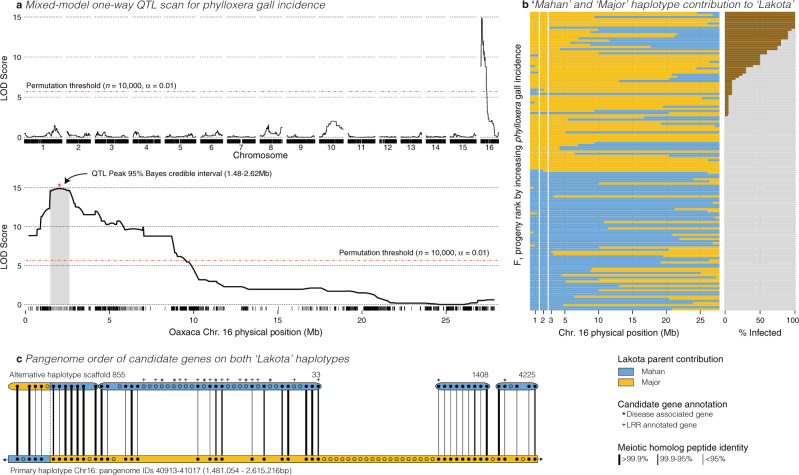
Genome-enabled biotechnologies have the potential to accelerate breeding efforts in long-lived perennial crop species. Despite the transformative potential of molecular tools in pecan and other outcrossing tree species, highly heterozygous genomes, significant presence-absence gene content variation, and histories of interspecific hybridization have constrained breeding efforts. To overcome these challenges, here, we present diploid genome assemblies and annotations of four outbred pecan genotypes, including a PacBio HiFi chromosome-scale assembly of both haplotypes of the 'Pawnee' cultivar. Comparative analysis and pan-genome integration reveal substantial and likely adaptive interspecific genomic introgressions, including an over-retained haplotype introgressed from bitternut hickory into pecan breeding pedigrees. Further, by leveraging our pan-genome presence-absence and functional annotation database among genomes and within the two outbred haplotypes of the 'Lakota' genome, we identify candidate genes for pest and pathogen resistance. Combined, these analyses and resources highlight significant progress towards functional and quantitative genomics in highly diverse and outbred crops.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
