

Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses
- PMID: 34226706
- PMCID: PMC8349845
- DOI: 10.1038/s41588-021-00892-1
Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses
Abstract
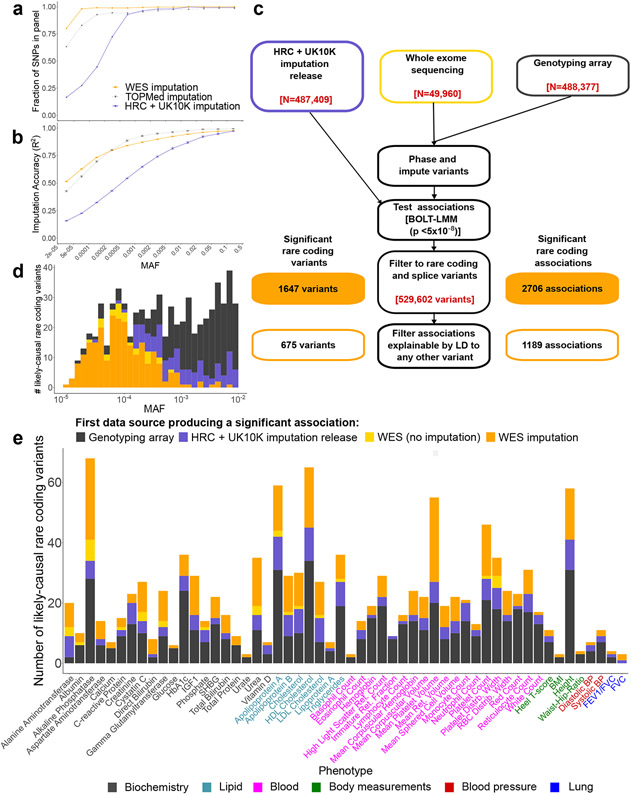
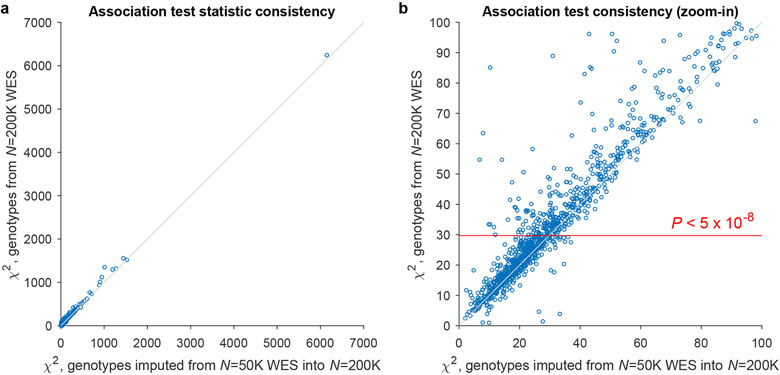
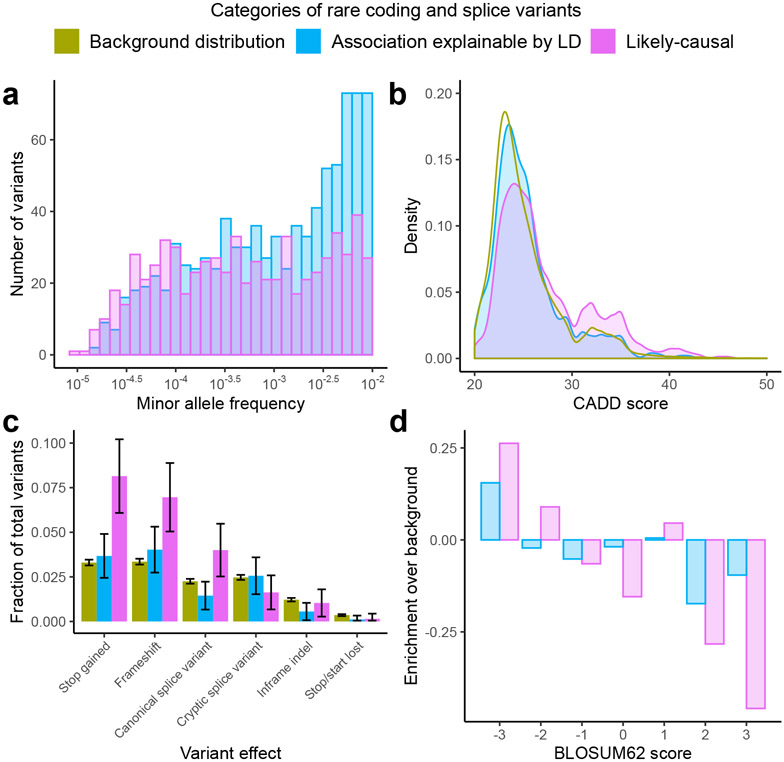
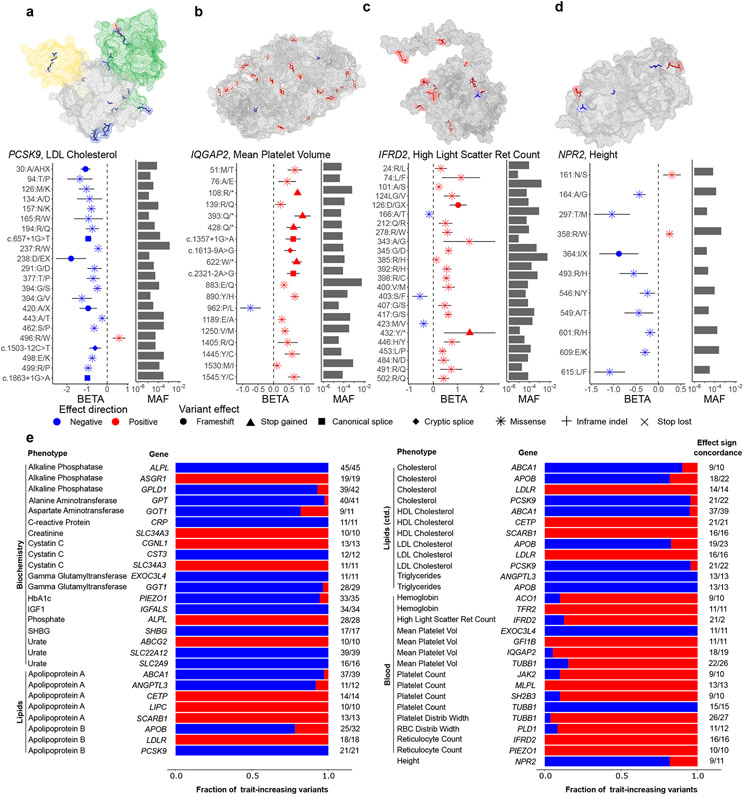
Exome association studies to date have generally been underpowered to systematically evaluate the phenotypic impact of very rare coding variants. We leveraged extensive haplotype sharing between 49,960 exome-sequenced UK Biobank participants and the remainder of the cohort (total n ≈ 500,000) to impute exome-wide variants with accuracy R2 > 0.5 down to minor allele frequency (MAF) ~0.00005. Association and fine-mapping analyses of 54 quantitative traits identified 1,189 significant associations (P < 5 × 10-8) involving 675 distinct rare protein-altering variants (MAF < 0.01) that passed stringent filters for likely causality. Across all traits, 49% of associations (578/1,189) occurred in genes with two or more hits; follow-up analyses of these genes identified allelic series containing up to 45 distinct 'likely-causal' variants. Our results demonstrate the utility of within-cohort imputation in population-scale genome-wide association studies, provide a catalog of likely-causal, large-effect coding variant associations and foreshadow the insights that will be revealed as genetic biobank studies continue to grow.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
METHODS REFERENCES
-
- Locke AE et al. Exome sequencing identifies high-impact trait-associated alleles enriched in Finns. bioRxiv 464255 (2019) doi:10.1101/464255. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
