

High-content single-cell combinatorial indexing
- PMID: 34226710
- PMCID: PMC8678206
- DOI: 10.1038/s41587-021-00962-z
High-content single-cell combinatorial indexing
Abstract
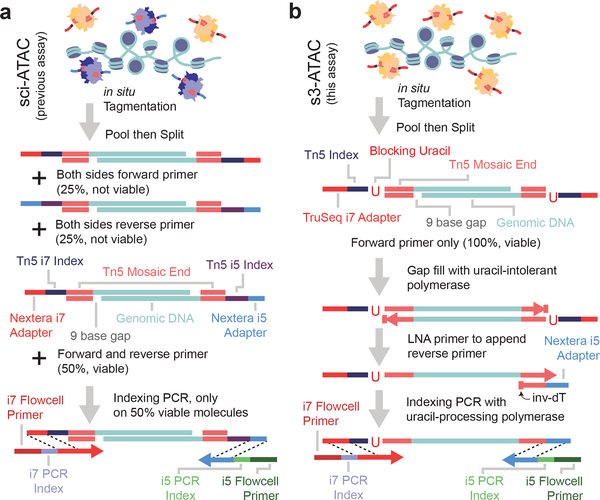
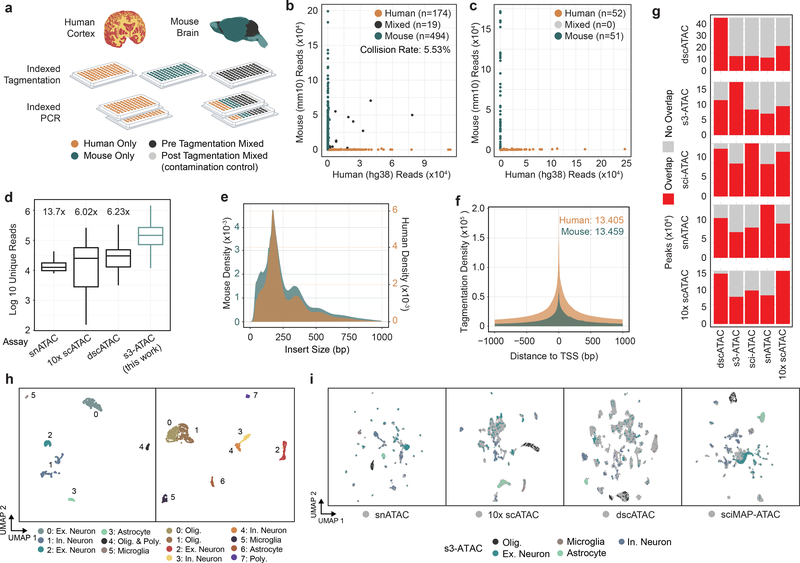
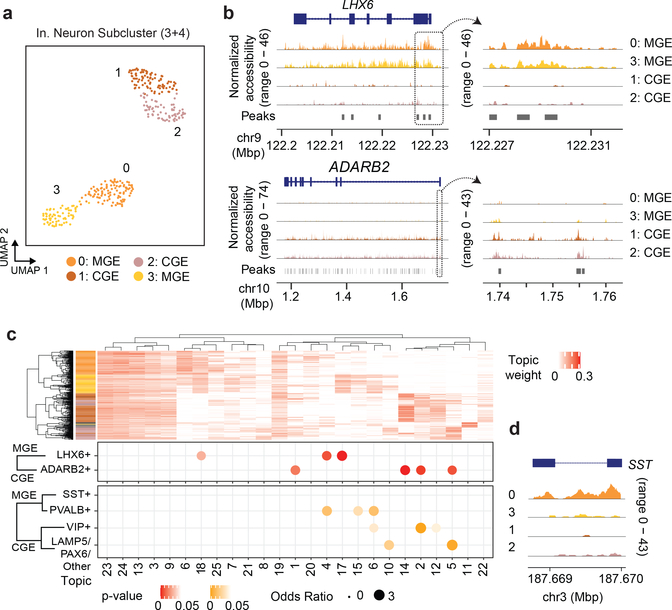
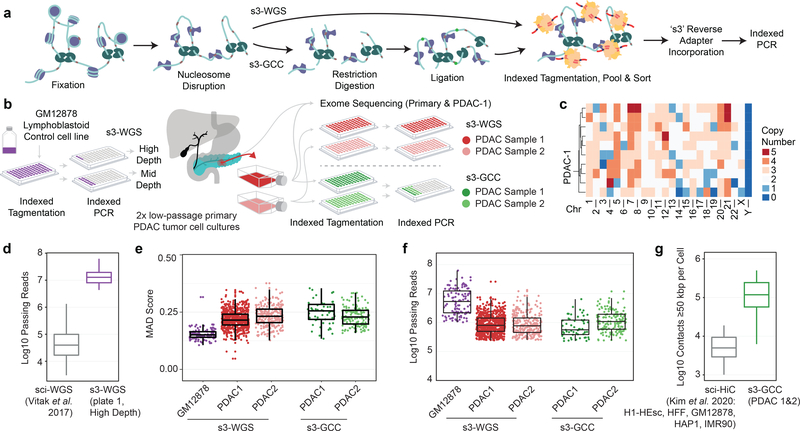
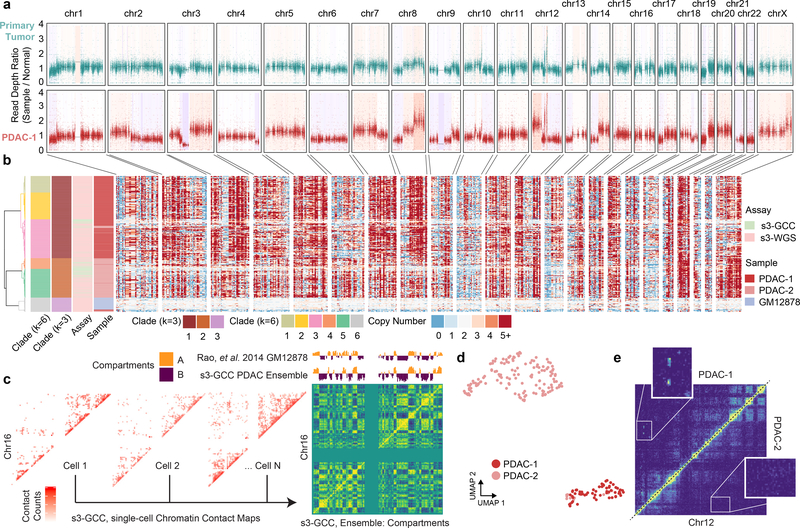
Single-cell combinatorial indexing (sci) with transposase-based library construction increases the throughput of single-cell genomics assays but produces sparse coverage in terms of usable reads per cell. We develop symmetrical strand sci ('s3'), a uracil-based adapter switching approach that improves the rate of conversion of source DNA into viable sequencing library fragments following tagmentation. We apply this chemistry to assay chromatin accessibility (s3-assay for transposase-accessible chromatin, s3-ATAC) in human cortical and mouse whole-brain tissues, with mouse datasets demonstrating a six- to 13-fold improvement in usable reads per cell compared with other available methods. Application of s3 to single-cell whole-genome sequencing (s3-WGS) and to whole-genome plus chromatin conformation (s3-GCC) yields 148- and 14.8-fold improvements, respectively, in usable reads per cell compared with sci-DNA-sequencing and sci-HiC. We show that s3-WGS and s3-GCC resolve subclonal genomic alterations in patient-derived pancreatic cancer cell lines. We expect that the s3 platform will be compatible with other transposase-based techniques, including sci-MET or CUT&Tag.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
Online Methods References
-
- Poplin R et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178 (2017) doi:10.1101/201178. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
