

Comparative genomic insights into the epidemiology and virulence of plant pathogenic pseudomonads from Turkey
- PMID: 34227931
- PMCID: PMC8477409
- DOI: 10.1099/mgen.0.000585
Comparative genomic insights into the epidemiology and virulence of plant pathogenic pseudomonads from Turkey
Abstract
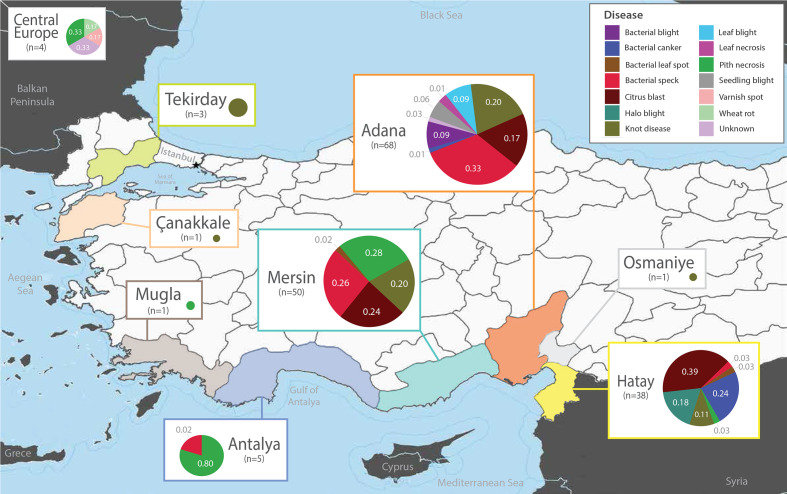
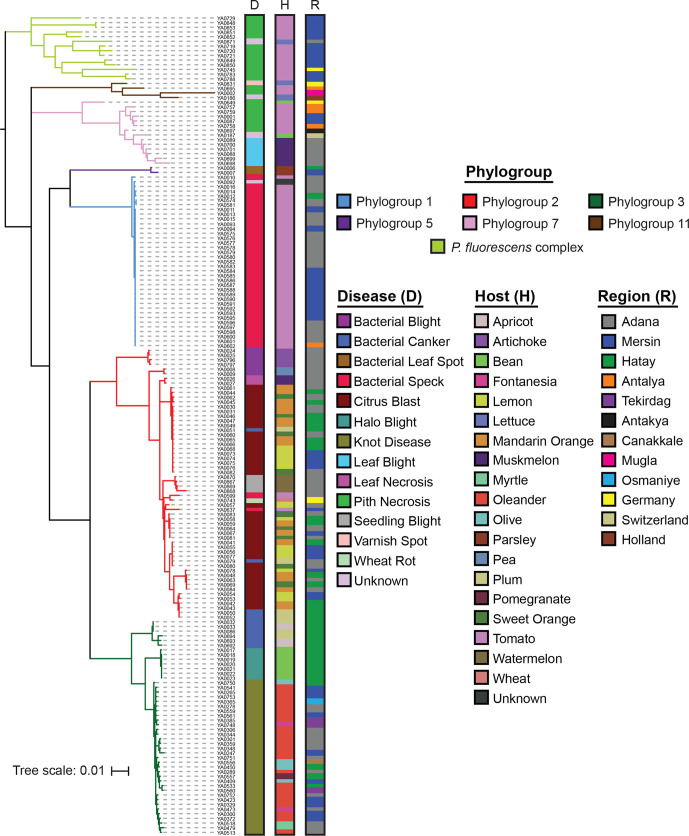
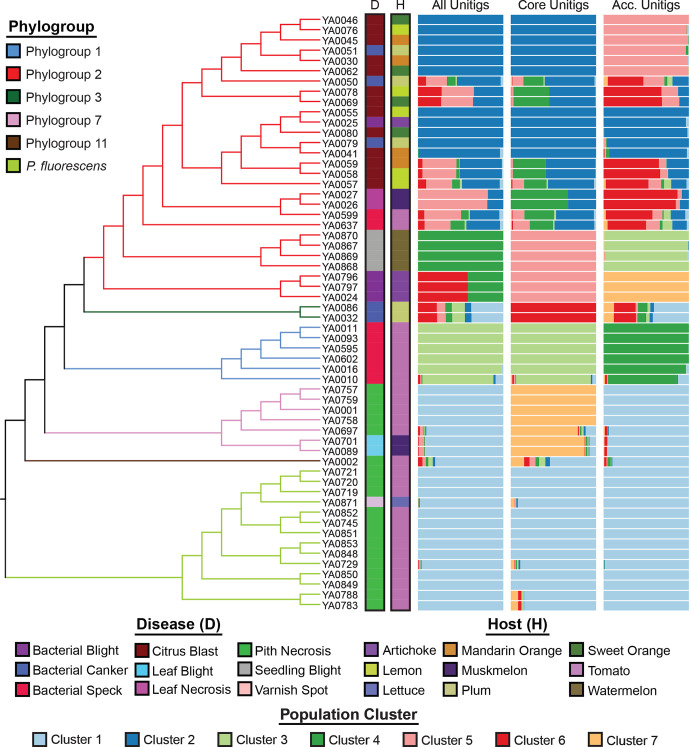
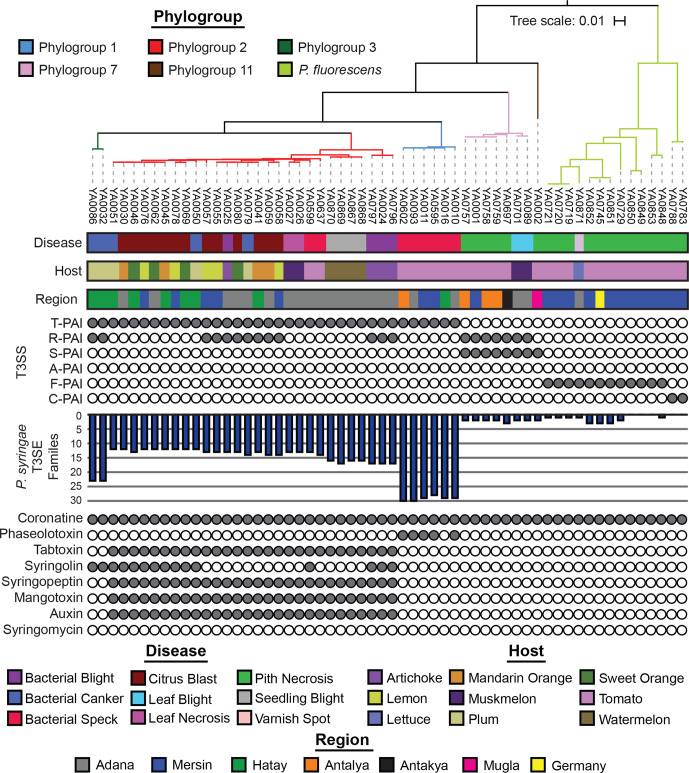
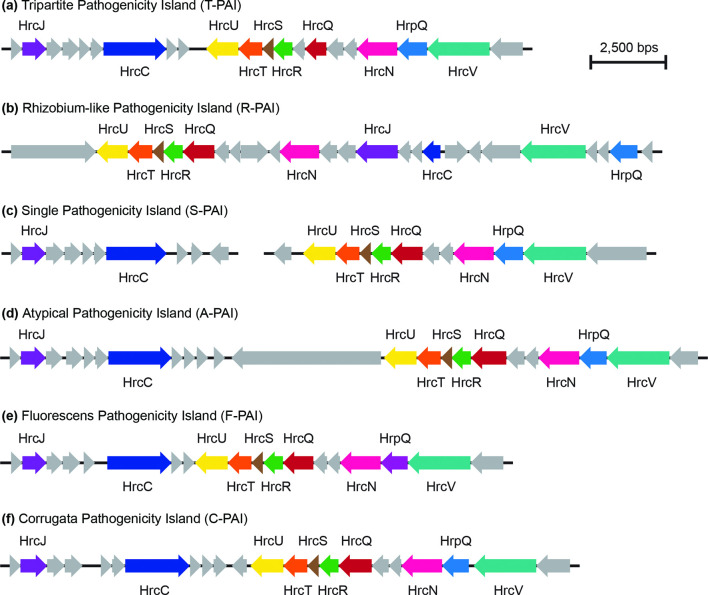
Pseudomonas is a highly diverse genus that includes species that cause disease in both plants and animals. Recently, pathogenic pseudomonads from the Pseudomonas syringae and Pseudomonas fluorescens species complexes have caused significant outbreaks in several agronomically important crops in Turkey, including tomato, citrus, artichoke and melon. We characterized 169 pathogenic Pseudomonas strains associated with recent outbreaks in Turkey via multilocus sequence analysis and whole-genome sequencing, then used comparative and evolutionary genomics to characterize putative virulence mechanisms. Most of the isolates are closely related to other plant pathogens distributed among the primary phylogroups of P. syringae, although there are significant numbers of P. fluorescens isolates, which is a species better known as a rhizosphere-inhabiting plant-growth promoter. We found that all 39 citrus blast pathogens cluster in P. syringae phylogroup 2, although strains isolated from the same host do not cluster monophyletically, with lemon, mandarin orange and sweet orange isolates all being intermixed throughout the phylogroup. In contrast, 20 tomato pith pathogens are found in two independent lineages: one in the P. syringae secondary phylogroups, and the other from the P. fluorescens species complex. These divergent pith necrosis strains lack characteristic virulence factors like the canonical tripartite type III secretion system, large effector repertoires and the ability to synthesize multiple bacterial phytotoxins, suggesting they have alternative molecular mechanisms to cause disease. These findings highlight the complex nature of host specificity among plant pathogenic pseudomonads.
Keywords: Pseudomonas fluorescens; Pseudomonas syringae; bacterial diseases; phytotoxins; plant pathogens; type III secreted effectors.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

Similar articles
-
Pathogenicity, phylogenomic, and comparative genomic study of Pseudomonas syringae sensu lato affecting sweet cherry in California.Microbiol Spectr. 2024 Oct 3;12(10):e0132424. doi: 10.1128/spectrum.01324-24. Epub 2024 Sep 3. Microbiol Spectr. 2024. PMID: 39225473 Free PMC article.
-
Comprehensive analysis of draft genomes of two closely related pseudomonas syringae phylogroup 2b strains infecting mono- and dicotyledon host plants.BMC Genomics. 2016 Dec 28;17(Suppl 14):1010. doi: 10.1186/s12864-016-3358-y. BMC Genomics. 2016. PMID: 28105943 Free PMC article.
-
Pseudomonas caspiana sp. nov., a citrus pathogen in the Pseudomonas syringae phylogenetic group.Syst Appl Microbiol. 2017 Jul;40(5):266-273. doi: 10.1016/j.syapm.2017.04.002. Epub 2017 May 11. Syst Appl Microbiol. 2017. PMID: 28552245
-
Evolution of plant pathogenesis in Pseudomonas syringae: a genomics perspective.Annu Rev Phytopathol. 2011;49:269-89. doi: 10.1146/annurev-phyto-072910-095242. Annu Rev Phytopathol. 2011. PMID: 21568703 Review.
-
Pseudomonas syringae type III secretion system effectors: repertoires in search of functions.Curr Opin Microbiol. 2009 Feb;12(1):53-60. doi: 10.1016/j.mib.2008.12.003. Epub 2009 Jan 23. Curr Opin Microbiol. 2009. PMID: 19168384 Review.
Cited by
-
Harnessing Pseudomonas spp. for sustainable plant crop protection.Front Microbiol. 2024 Nov 21;15:1485197. doi: 10.3389/fmicb.2024.1485197. eCollection 2024. Front Microbiol. 2024. PMID: 39640850 Free PMC article. Review.
References
-
- Lamichhane JR, Messéan A, Morris CE. Insights into epidemiology and control of diseases of annual plants caused by the Pseudomonas syringae species complex. J Gen Plant Pathol. 2015;81:331–350. doi: 10.1007/s10327-015-0605-z. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials