

Infectious disease-associated encephalopathies
- PMID: 34229735
- PMCID: PMC8259088
- DOI: 10.1186/s13054-021-03659-6
Infectious disease-associated encephalopathies
Abstract
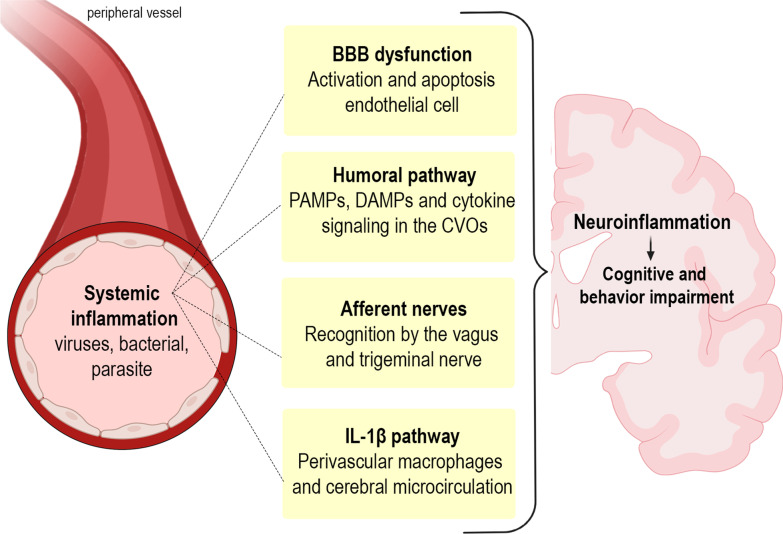
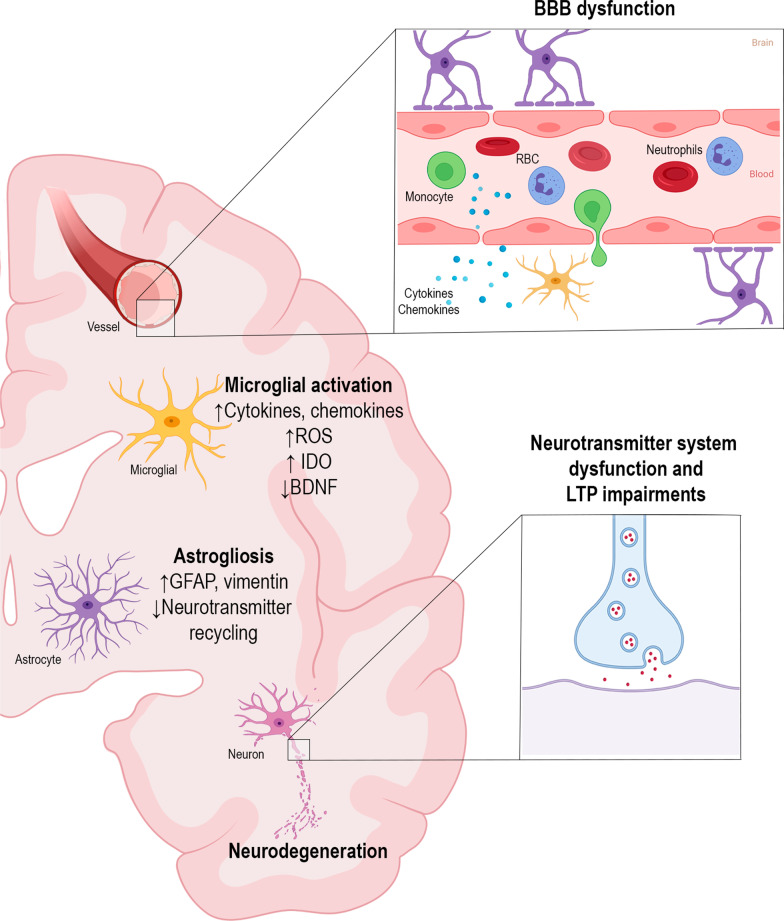
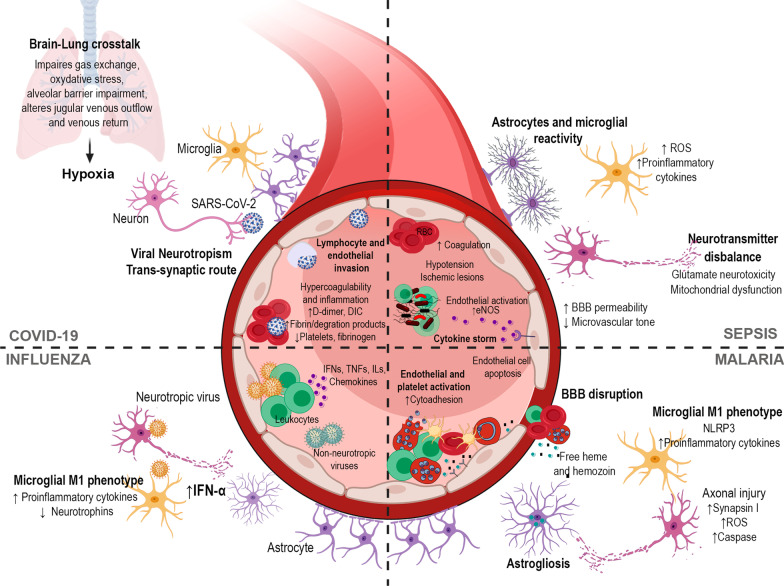
Infectious diseases may affect brain function and cause encephalopathy even when the pathogen does not directly infect the central nervous system, known as infectious disease-associated encephalopathy. The systemic inflammatory process may result in neuroinflammation, with glial cell activation and increased levels of cytokines, reduced neurotrophic factors, blood-brain barrier dysfunction, neurotransmitter metabolism imbalances, and neurotoxicity, and behavioral and cognitive impairments often occur in the late course. Even though infectious disease-associated encephalopathies may cause devastating neurologic and cognitive deficits, the concept of infectious disease-associated encephalopathies is still under-investigated; knowledge of the underlying mechanisms, which may be distinct from those of encephalopathies of non-infectious cause, is still limited. In this review, we focus on the pathophysiology of encephalopathies associated with peripheral (sepsis, malaria, influenza, and COVID-19), emerging therapeutic strategies, and the role of neuroinflammation.
Keywords: COVID-19; Cognition; Encephalopathy; Infection; Influenza; Malaria; Microglial priming; Neuroinflammation; SARS-CoV-2; Sepsis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
