

Human deafness-associated variants alter the dynamics of key molecules in hair cell stereocilia F-actin cores
- PMID: 34232383
- PMCID: PMC11351816
- DOI: 10.1007/s00439-021-02304-0
Human deafness-associated variants alter the dynamics of key molecules in hair cell stereocilia F-actin cores
Abstract
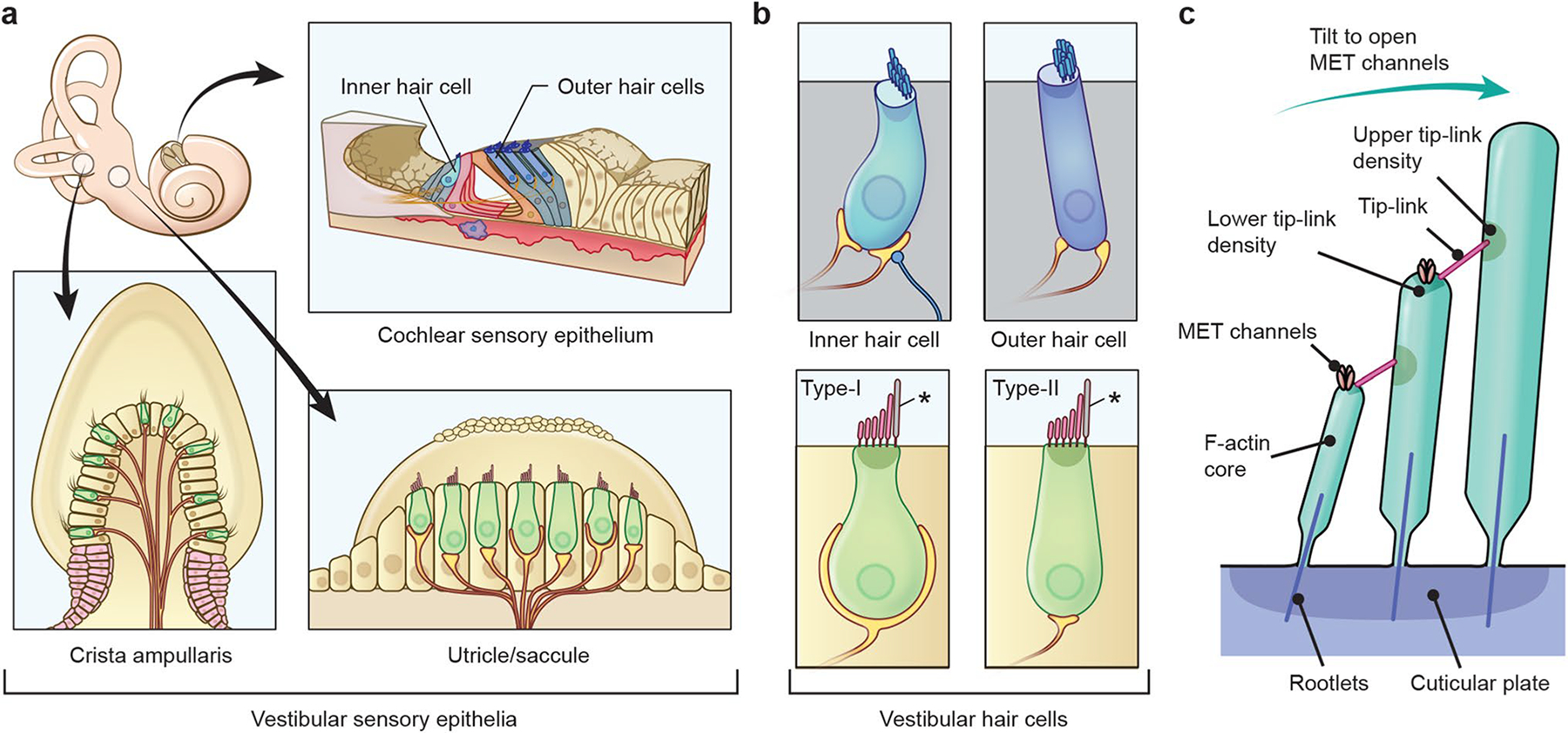
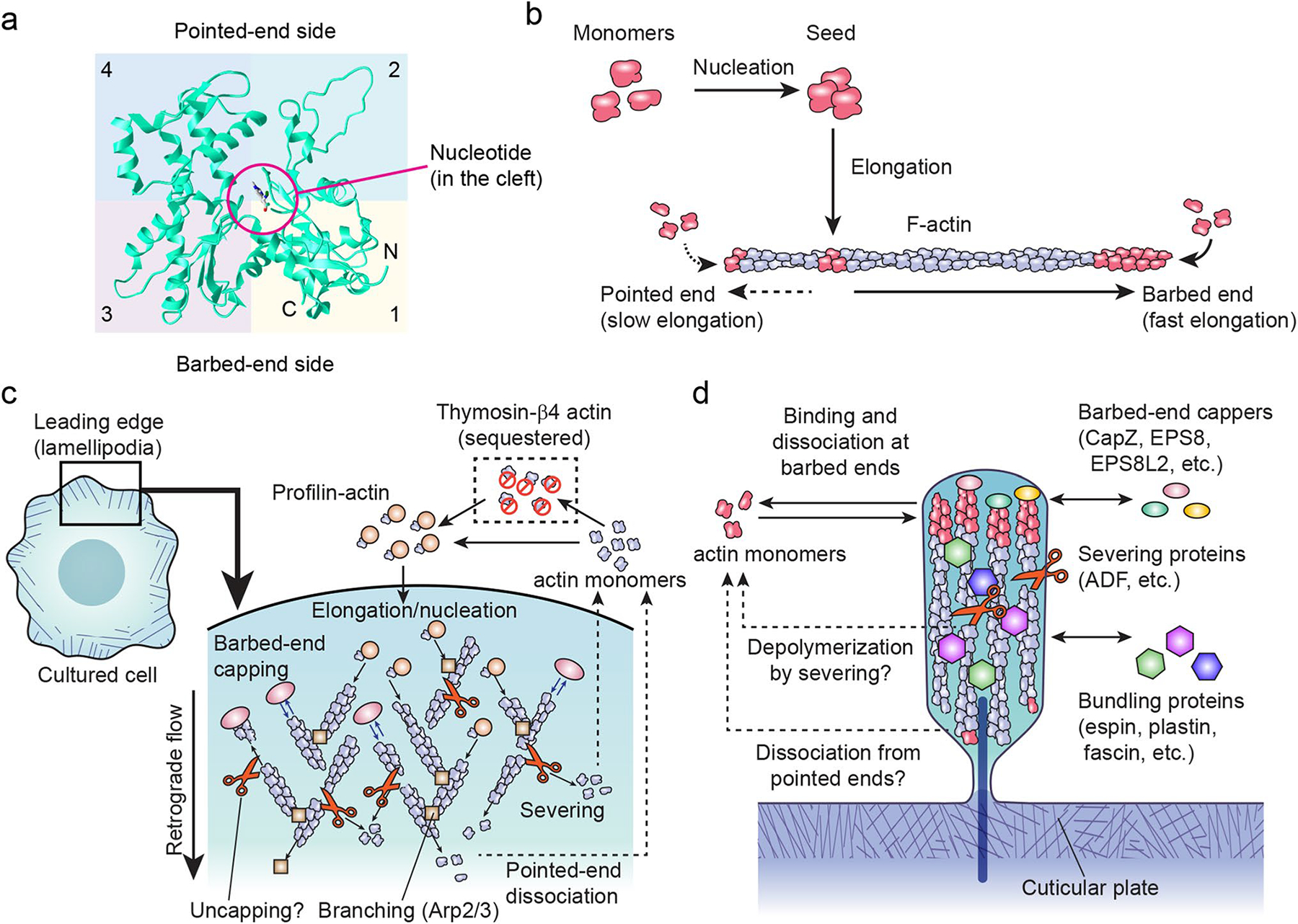
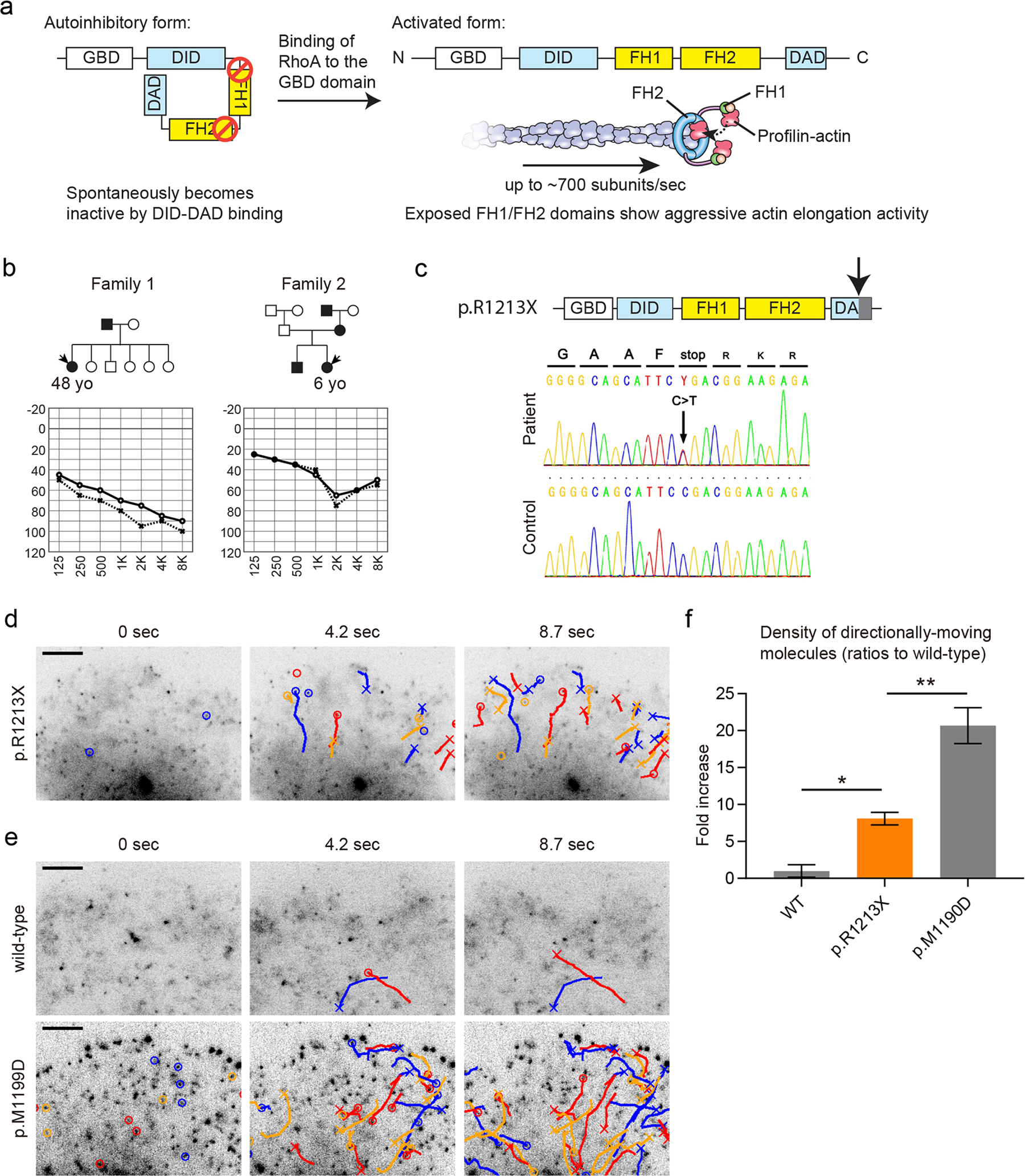
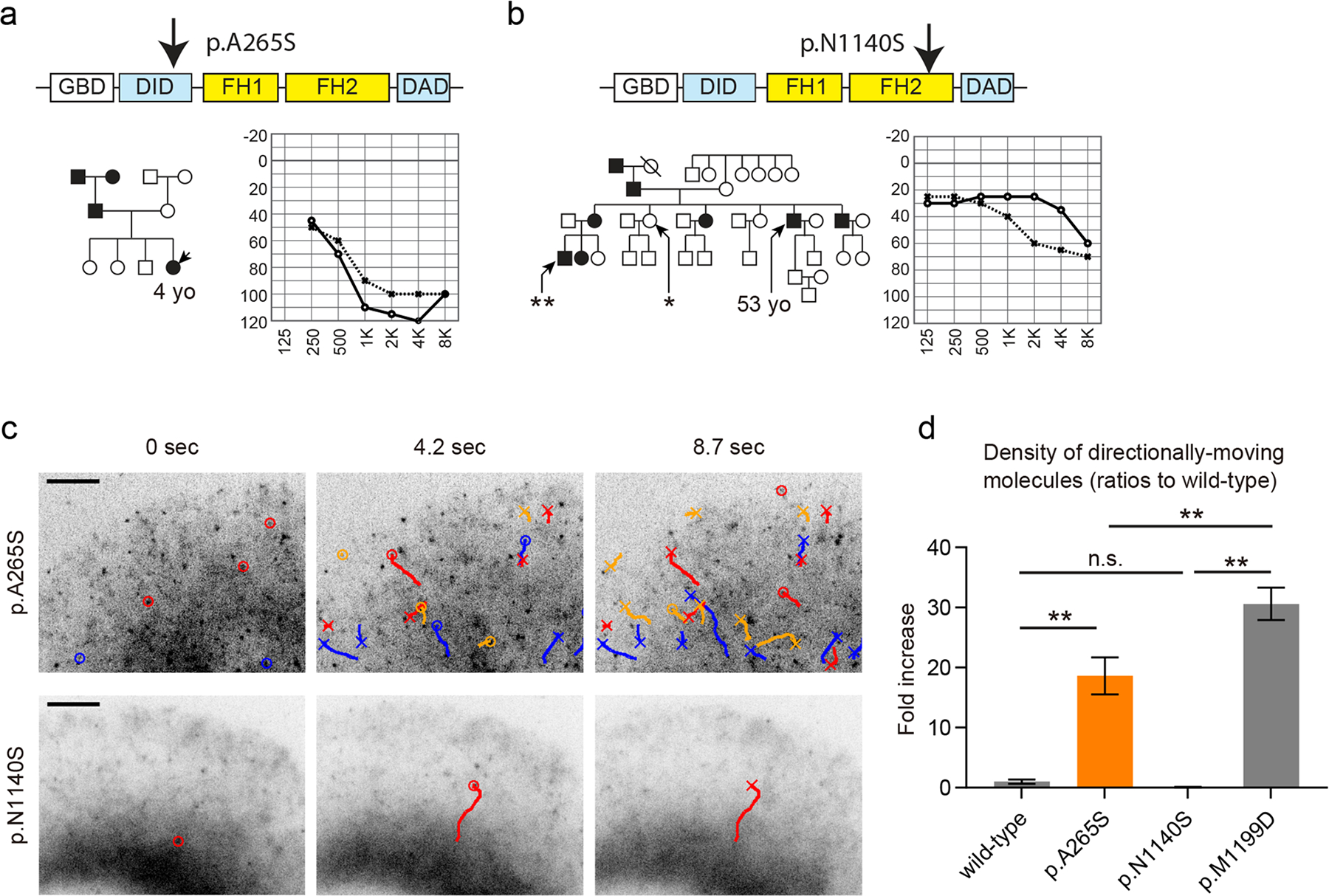
Stereocilia protrude up to 100 µm from the apical surface of vertebrate inner ear hair cells and are packed with cross-linked filamentous actin (F-actin). They function as mechanical switches to convert sound vibration into electrochemical neuronal signals transmitted to the brain. Several genes encode molecular components of stereocilia including actin monomers, actin regulatory and bundling proteins, motor proteins and the proteins of the mechanotransduction complex. A stereocilium F-actin core is a dynamic system, which is continuously being remodeled while maintaining an outwardly stable architecture under the regulation of F-actin barbed-end cappers, severing proteins and crosslinkers. The F-actin cores of stereocilia also provide a pathway for motor proteins to transport cargos including components of tip-link densities, scaffolding proteins and actin regulatory proteins. Deficiencies and mutations of stereocilia components that disturb this "dynamic equilibrium" in stereocilia can induce morphological changes and disrupt mechanotransduction causing sensorineural hearing loss, best studied in mouse and zebrafish models. Currently, at least 23 genes, associated with human syndromic and nonsyndromic hearing loss, encode proteins involved in the development and maintenance of stereocilia F-actin cores. However, it is challenging to predict how variants associated with sensorineural hearing loss segregating in families affect protein function. Here, we review the functions of several molecular components of stereocilia F-actin cores and provide new data from our experimental approach to directly evaluate the pathogenicity and functional impact of reported and novel variants of DIAPH1 in autosomal-dominant DFNA1 hearing loss using single-molecule fluorescence microscopy.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Declarations
Figures
References
-
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PS, Deshmukh D, Griffith AJ, Riazuddin S, Friedman TB, Wilcox ER (2002) Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet 110:527–531. 10.1007/s00439-002-0732-4 - DOI - PubMed
-
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Riazuddin S, Griffith AJ, Friedman TB, Wilcox ER (2003a) Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet 72:1315–1322. 10.1086/375122 - DOI - PMC - PubMed
-
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER (2003b) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12:3215–3223. 10.1093/hmg/ddg358 - DOI - PubMed
-
- Al-Maawali A, Barry BJ, Rajab A, El-Quessny M, Seman A, Coury SN, Barkovich AJ, Yang E, Walsh CA, Mochida GH, Stoler JM (2016) Novel loss-of-function variants in DIAPH1 associated with syndromic microcephaly, blindness, and early onset seizures. Am J Med Genet A 170A:435–440. 10.1002/ajmg.a.37422 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
