

In-Cell Labeling and Mass Spectrometry for Systems-Level Structural Biology
- PMID: 34232610
- PMCID: PMC8966414
- DOI: 10.1021/acs.chemrev.1c00223
In-Cell Labeling and Mass Spectrometry for Systems-Level Structural Biology
Abstract
Biological systems have evolved to utilize proteins to accomplish nearly all functional roles needed to sustain life. A majority of biological functions occur within the crowded environment inside cells and subcellular compartments where proteins exist in a densely packed complex network of protein-protein interactions. The structural biology field has experienced a renaissance with recent advances in crystallography, NMR, and CryoEM that now produce stunning models of large and complex structures previously unimaginable. Nevertheless, measurements of such structural detail within cellular environments remain elusive. This review will highlight how advances in mass spectrometry, chemical labeling, and informatics capabilities are merging to provide structural insights on proteins, complexes, and networks that exist inside cells. Because of the molecular detection specificity provided by mass spectrometry and proteomics, these approaches provide systems-level information that not only benefits from conventional structural analysis, but also is highly complementary. Although far from comprehensive in their current form, these approaches are currently providing systems structural biology information that can uniquely reveal how conformations and interactions involving many proteins change inside cells with perturbations such as disease, drug treatment, or phenotypic differences. With continued advancements and more widespread adaptation, systems structural biology based on in-cell labeling and mass spectrometry will provide an even greater wealth of structural knowledge.
Figures




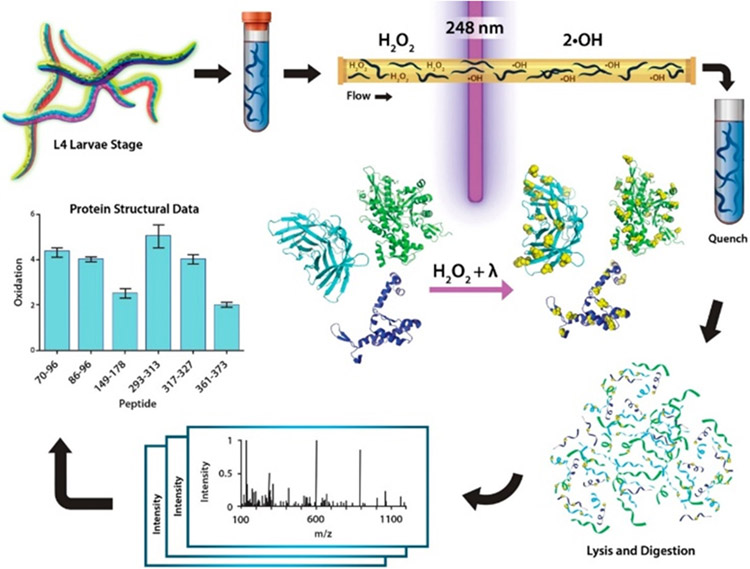
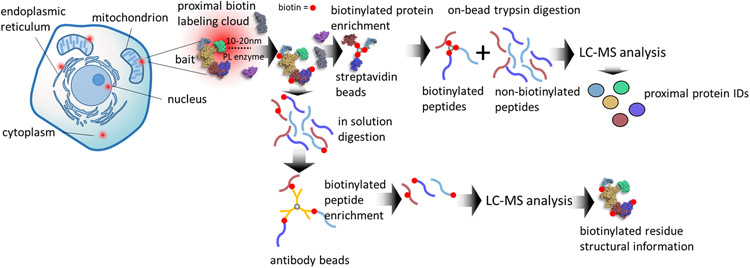































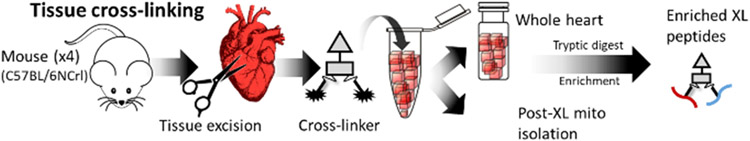


Similar articles
-
Surface induced dissociation: dissecting noncovalent protein complexes in the gas phase.Acc Chem Res. 2014 Apr 15;47(4):1010-8. doi: 10.1021/ar400223t. Epub 2014 Feb 13. Acc Chem Res. 2014. PMID: 24524650
-
Towards integrative structural mass spectrometry: Benefits from hybrid approaches.Methods. 2015 Nov 1;89:4-12. doi: 10.1016/j.ymeth.2015.05.024. Epub 2015 May 29. Methods. 2015. PMID: 26028598 Review.
-
Crosslinking mass spectrometry: A link between structural biology and systems biology.Protein Sci. 2021 Apr;30(4):773-784. doi: 10.1002/pro.4045. Epub 2021 Mar 6. Protein Sci. 2021. PMID: 33594738 Free PMC article. Review.
-
Chemical cross-linking with mass spectrometry: a tool for systems structural biology.Curr Opin Chem Biol. 2019 Feb;48:8-18. doi: 10.1016/j.cbpa.2018.08.006. Epub 2018 Aug 30. Curr Opin Chem Biol. 2019. PMID: 30172868 Free PMC article. Review.
-
Joining forces: integrating proteomics and cross-linking with the mass spectrometry of intact complexes.Mol Cell Proteomics. 2012 Mar;11(3):R111.014027. doi: 10.1074/mcp.R111.014027. Epub 2011 Dec 16. Mol Cell Proteomics. 2012. PMID: 22180098 Free PMC article. Review.
Cited by
-
Exploring an Alternative Cysteine-Reactive Chemistry to Enable Proteome-Wide PPI Analysis by Cross-Linking Mass Spectrometry.Anal Chem. 2023 Jan 31;95(4):2532-2539. doi: 10.1021/acs.analchem.2c04986. Epub 2023 Jan 18. Anal Chem. 2023. PMID: 36652389 Free PMC article.
-
Precision Targeting of Endogenous Epidermal Growth Factor Receptor (EGFR) by Structurally Aligned Dual-Modifier Labeling.ACS Pharmacol Transl Sci. 2022 Sep 19;5(10):859-871. doi: 10.1021/acsptsci.2c00155. eCollection 2022 Oct 14. ACS Pharmacol Transl Sci. 2022. PMID: 36268127 Free PMC article.
-
Mass Spectrometry-Based Proteomics Technologies to Define Endogenous Protein-Protein Interactions and Their Applications to Cancer and Viral Infectious Diseases.Mass Spectrom Rev. 2025 Feb 9:10.1002/mas.21926. doi: 10.1002/mas.21926. Online ahead of print. Mass Spectrom Rev. 2025. PMID: 39924651 Free PMC article. Review.
-
Using mass spectrometry-based methods to understand amyloid formation and inhibition of alpha-synuclein and amyloid beta.Mass Spectrom Rev. 2024 Jul-Aug;43(4):782-825. doi: 10.1002/mas.21814. Epub 2022 Oct 12. Mass Spectrom Rev. 2024. PMID: 36224716 Free PMC article. Review.
-
Dissecting the structural heterogeneity of proteins by native mass spectrometry.Protein Sci. 2023 Apr;32(4):e4612. doi: 10.1002/pro.4612. Protein Sci. 2023. PMID: 36851867 Free PMC article. Review.
References
-
- Ellis RJ Macromolecular Crowding: Obvious but Underappreciated. Trends in Biochemical Sciences 2001, 26, 597–604. - PubMed
-
- Stadmiller SS; Aguilar JS; Parnham S; Pielak GJ Protein–Peptide Binding Energetics under Crowded Conditions. J. Phys. Chem. B 2020, 124, 9297–9309. - PubMed
-
- Gruebele M; Pielak GJ Dynamical Spectroscopy and Microscopy of Proteins in Cells. Current Opinion in Structural Biology 2021, 70, 1–7. - PubMed
-
- Sali A; Glaeser R; Earnest T; Baumeister W From Words to Literature in Structural Proteomics. Nature 2003, 422, 216–225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
