

CARD9 mediates glucose-stimulated insulin secretion in pancreatic beta cells
- PMID: 34233162
- PMCID: PMC8478856
- DOI: 10.1016/j.bcp.2021.114670
CARD9 mediates glucose-stimulated insulin secretion in pancreatic beta cells
Abstract
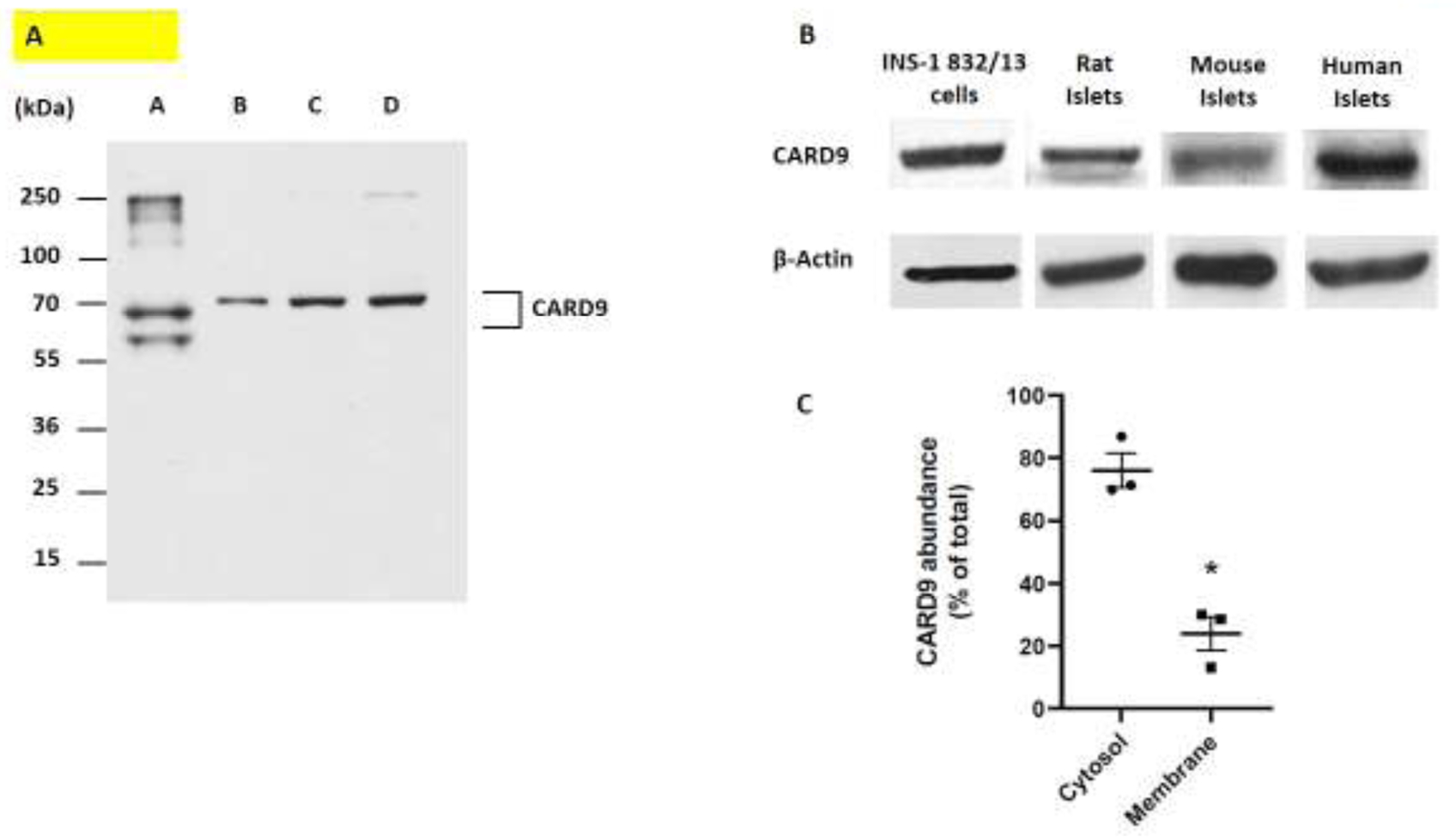
Caspase recruitment domain containing protein 9 (CARD9) plays key regulatory role(s) in innate and adaptive immune responses. Recent evidence implicates CARD9 in the onset of metabolic diseases including insulin resistance. However, potential contributory roles of CARD9 in glucose-stimulated insulin secretion (GSIS) remain unknown. Herein, we report that CARD9 is expressed in human islets, rat islets, mouse islets and clonal INS-1 832/13 cells. Subcellularly, CARD9 is predominantly cytosolic (~75%) in INS-1 832/13 cells. siRNA-mediated depletion of CARD9 expression significantly (~50%) suppressed GSIS in INS-1 832/13 cells. Interestingly, glucose-induced activation of Rac1, a small G-protein, which is a requisite for GSIS to occur, is unaffected in CARD9-si transfected cells, suggesting that CARD9-mediates GSIS in a Rac1-independent fashion. Furthermore, insulin secretion promoted by KCl or mastoparan (a global G protein activator), remained resistant to CARD9 depletion in INS-1 832/13 cells. In addition, pharmacological inhibition (BRD5529) of interaction between CARD9 and TRIM62, its ubiquitin ligase, exerted no significant effects on GSIS. Lastly, depletion of CARD9 prevented glucose-induced p38, not ERK1/2 phosphorylation in beta cells. Based on these observations, we propose that CARD9 might regulate GSIS via a Rac1-independent and p38-dependent signaling module.
Keywords: CARD9; Insulin secretion; Pancreatic beta cell; Rac1.
Published by Elsevier Inc.
Conflict of interest statement
Conflict of Interests
The authors declare no conflict of interests
Figures







Similar articles
-
CARD9 Mediates Pancreatic Islet Beta-Cell Dysfunction Under the Duress of Hyperglycemic Stress.Cell Physiol Biochem. 2022 Apr 1;56(2):120-137. doi: 10.33594/000000508. Cell Physiol Biochem. 2022. PMID: 35362297 Free PMC article.
-
Regulatory roles of CARD9-BCL10-Rac1 (CBR) signalome in islet β-cell function in health and metabolic stress: Is there room for MALT1?Biochem Pharmacol. 2023 Dec;218:115889. doi: 10.1016/j.bcp.2023.115889. Epub 2023 Oct 29. Biochem Pharmacol. 2023. PMID: 37991197 Free PMC article. Review.
-
Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 β-cells and rat islets.Biochem Pharmacol. 2011 Apr 15;81(8):1016-27. doi: 10.1016/j.bcp.2011.01.006. Epub 2011 Jan 26. Biochem Pharmacol. 2011. PMID: 21276423 Free PMC article.
-
VAV2, a guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta cells.Diabetologia. 2015 Nov;58(11):2573-81. doi: 10.1007/s00125-015-3707-4. Epub 2015 Jul 31. Diabetologia. 2015. PMID: 26224100 Free PMC article.
-
Protein histidine [de]phosphorylation in insulin secretion: abnormalities in models of impaired insulin secretion.Naunyn Schmiedebergs Arch Pharmacol. 2011 Oct;384(4-5):383-90. doi: 10.1007/s00210-011-0616-z. Epub 2011 Apr 6. Naunyn Schmiedebergs Arch Pharmacol. 2011. PMID: 21626002 Free PMC article. Review.
Cited by
-
The RhoGDIβ-Rac1-CARD9 Signaling Module Mediates Islet β-Cell Dysfunction Under Chronic Hyperglycemia.Cells. 2025 Jul 9;14(14):1046. doi: 10.3390/cells14141046. Cells. 2025. PMID: 40710299 Free PMC article. Review.
-
Regulatory Roles of Histone Deacetylation in Metabolic Stress-Induced Expression of Caspase Recruitment Domain-Containing Protein 9 (CARD9) in Pancreatic β-Cells.Int J Mol Sci. 2023 Nov 6;24(21):15994. doi: 10.3390/ijms242115994. Int J Mol Sci. 2023. PMID: 37958977 Free PMC article.
-
CARD9 Mediates Pancreatic Islet Beta-Cell Dysfunction Under the Duress of Hyperglycemic Stress.Cell Physiol Biochem. 2022 Apr 1;56(2):120-137. doi: 10.33594/000000508. Cell Physiol Biochem. 2022. PMID: 35362297 Free PMC article.
-
Regulatory roles of CARD9-BCL10-Rac1 (CBR) signalome in islet β-cell function in health and metabolic stress: Is there room for MALT1?Biochem Pharmacol. 2023 Dec;218:115889. doi: 10.1016/j.bcp.2023.115889. Epub 2023 Oct 29. Biochem Pharmacol. 2023. PMID: 37991197 Free PMC article. Review.
References
-
- MacDonald MJ, Elusive proximal signals of beta-cells for insulin secretion, Diabetes 39(12) (1990) 1461–6. - PubMed
-
- Newgard CB, McGarry JD, Metabolic coupling factors in pancreatic beta-cell signal transduction, Annu Rev Biochem 64 (1995) 689–719. - PubMed
-
- Prentki M, Peyot ML, Masiello P, Madiraju SRM, Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell, Diabetes 69(3) (2020) 279–290. - PubMed
-
- Maechler P, Mitochondrial function and insulin secretion, Mol Cell Endocrinol 379(1–2) (2013) 12–8. - PubMed
-
- Tengholm A, Gylfe E, cAMP signalling in insulin and glucagon secretion, Diabetes Obes Metab 19Suppl 1 (2017) 42–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
