

The signal pathways and treatment of cytokine storm in COVID-19
- PMID: 34234112
- PMCID: PMC8261820
- DOI: 10.1038/s41392-021-00679-0
The signal pathways and treatment of cytokine storm in COVID-19
Erratum in
-
Correction: The signal pathways and treatment of cytokine storm in COVID-19.Signal Transduct Target Ther. 2021 Aug 31;6(1):326. doi: 10.1038/s41392-021-00744-8. Signal Transduct Target Ther. 2021. PMID: 34465720 Free PMC article. No abstract available.
Abstract
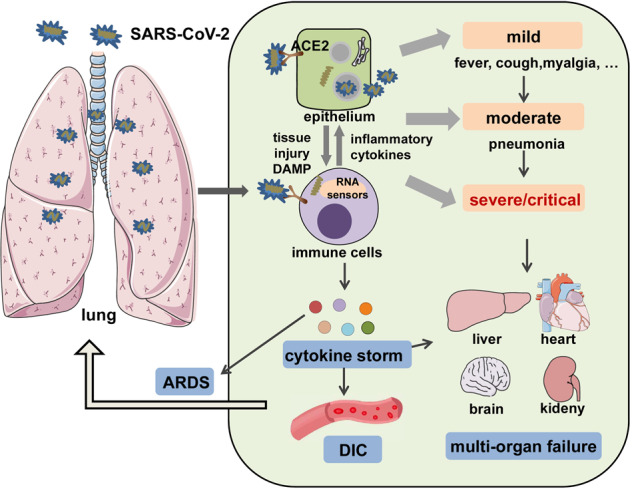
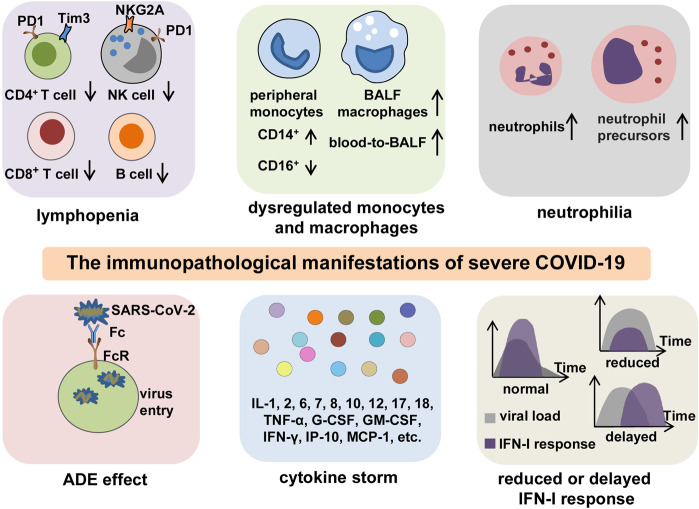
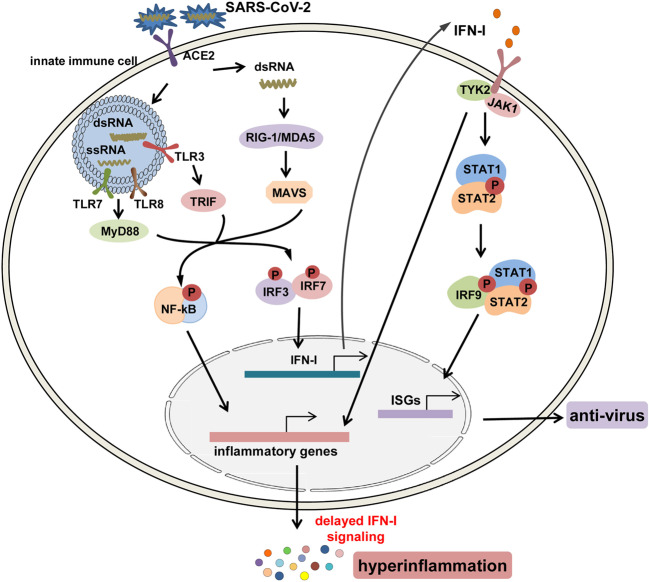
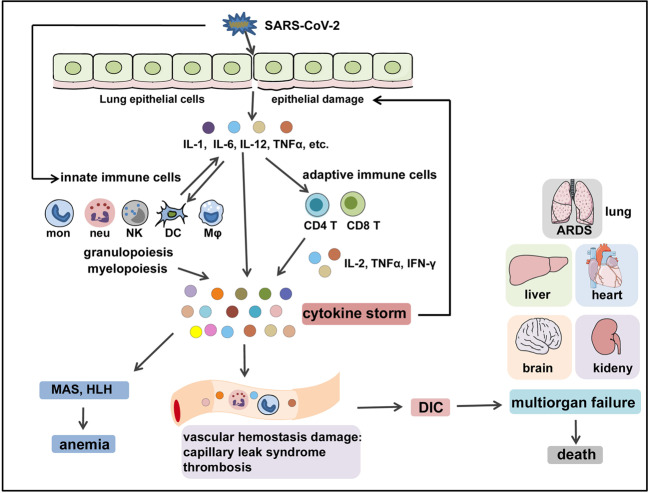
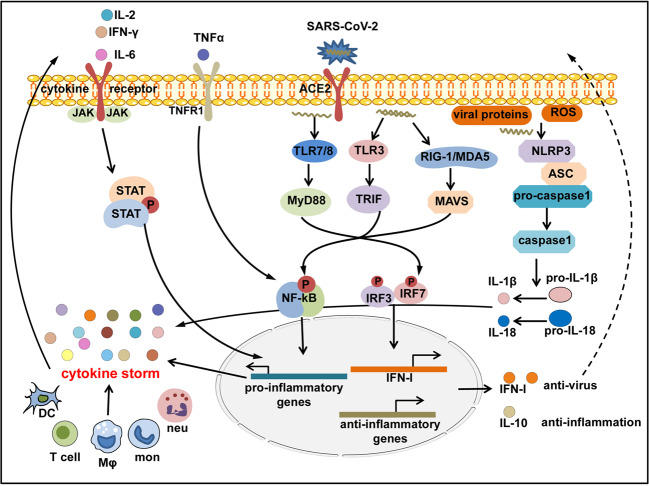
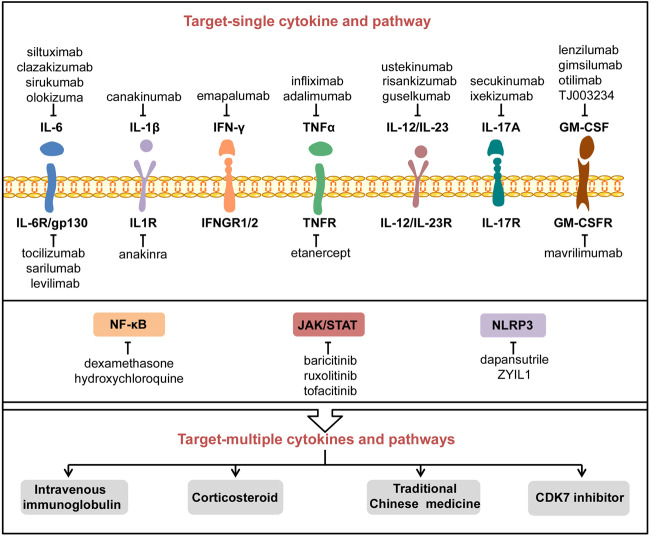
The Coronavirus Disease 2019 (COVID-19) pandemic has become a global crisis and is more devastating than any other previous infectious disease. It has affected a significant proportion of the global population both physically and mentally, and destroyed businesses and societies. Current evidence suggested that immunopathology may be responsible for COVID-19 pathogenesis, including lymphopenia, neutrophilia, dysregulation of monocytes and macrophages, reduced or delayed type I interferon (IFN-I) response, antibody-dependent enhancement, and especially, cytokine storm (CS). The CS is characterized by hyperproduction of an array of pro-inflammatory cytokines and is closely associated with poor prognosis. These excessively secreted pro-inflammatory cytokines initiate different inflammatory signaling pathways via their receptors on immune and tissue cells, resulting in complicated medical symptoms including fever, capillary leak syndrome, disseminated intravascular coagulation, acute respiratory distress syndrome, and multiorgan failure, ultimately leading to death in the most severe cases. Therefore, it is clinically important to understand the initiation and signaling pathways of CS to develop more effective treatment strategies for COVID-19. Herein, we discuss the latest developments in the immunopathological characteristics of COVID-19 and focus on CS including the current research status of the different cytokines involved. We also discuss the induction, function, downstream signaling, and existing and potential interventions for targeting these cytokines or related signal pathways. We believe that a comprehensive understanding of CS in COVID-19 will help to develop better strategies to effectively control immunopathology in this disease and other infectious and inflammatory diseases.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 81974248/National Natural Science Foundation of China (National Science Foundation of China)
- 81671561/National Natural Science Foundation of China (National Science Foundation of China)
- 81671561/National Natural Science Foundation of China (National Science Foundation of China)
- 81974248/National Natural Science Foundation of China (National Science Foundation of China)
- 81974248/National Natural Science Foundation of China (National Science Foundation of China)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
