

Genomic epidemiology of SARS-CoV-2 in the UAE reveals novel virus mutation, patterns of co-infection and tissue specific host immune response
- PMID: 34234167
- PMCID: PMC8263779
- DOI: 10.1038/s41598-021-92851-3
Genomic epidemiology of SARS-CoV-2 in the UAE reveals novel virus mutation, patterns of co-infection and tissue specific host immune response
Erratum in
-
Publisher Correction: Genomic epidemiology of SARS-CoV-2 in the UAE reveals novel virus mutation, patterns of co-infection and tissue specific host immune response.Sci Rep. 2021 Sep 15;11(1):18728. doi: 10.1038/s41598-021-98373-2. Sci Rep. 2021. PMID: 34526633 Free PMC article. No abstract available.
Abstract
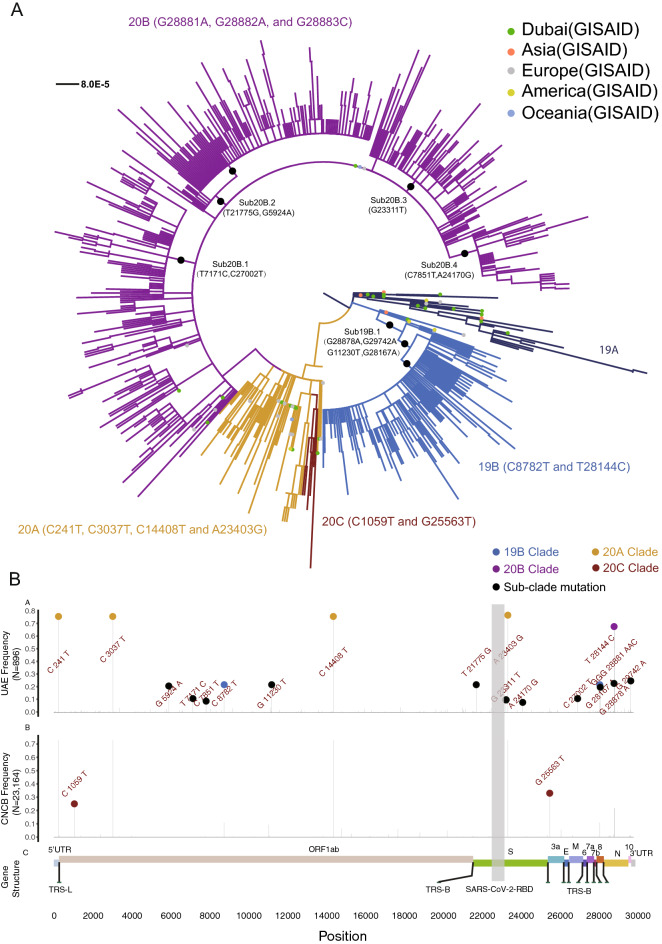
To unravel the source of SARS-CoV-2 introduction and the pattern of its spreading and evolution in the United Arab Emirates, we conducted meta-transcriptome sequencing of 1067 nasopharyngeal swab samples collected between May 9th and Jun 29th, 2020 during the first peak of the local COVID-19 epidemic. We identified global clade distribution and eleven novel genetic variants that were almost absent in the rest of the world and that defined five subclades specific to the UAE viral population. Cross-settlement human-to-human transmission was related to the local business activity. Perhaps surprisingly, at least 5% of the population were co-infected by SARS-CoV-2 of multiple clades within the same host. We also discovered an enrichment of cytosine-to-uracil mutation among the viral population collected from the nasopharynx, that is different from the adenosine-to-inosine change previously reported in the bronchoalveolar lavage fluid samples and a previously unidentified upregulation of APOBEC4 expression in nasopharynx among infected patients, indicating the innate immune host response mediated by ADAR and APOBEC gene families could be tissue-specific. The genomic epidemiological and molecular biological knowledge reported here provides new insights for the SARS-CoV-2 evolution and transmission and points out future direction on host-pathogen interaction investigation.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC, Du B, Li L, Zeng G, Yuen KY, Chen R, Tang C, Wang T, Chen P, Xiang J, Li S, Wang JL, Liang Z, Peng Y, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu J, Chen Z, Li G, Zheng Z, Qiu S, Luo J, Ye C, Zhu S, Zhong N. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2002032. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
