

Mapping the human genetic architecture of COVID-19
- PMID: 34237774
- PMCID: PMC8674144
- DOI: 10.1038/s41586-021-03767-x
Mapping the human genetic architecture of COVID-19
Abstract
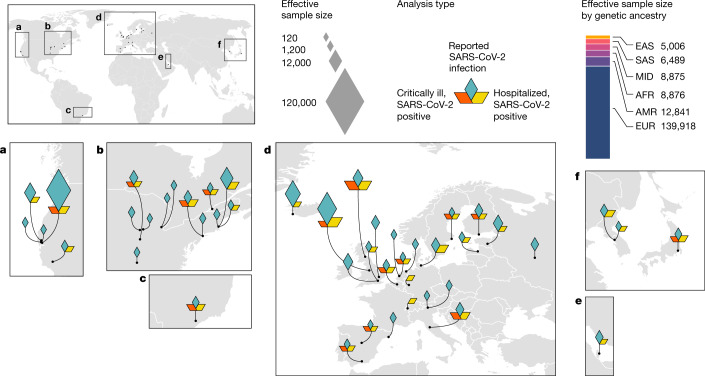
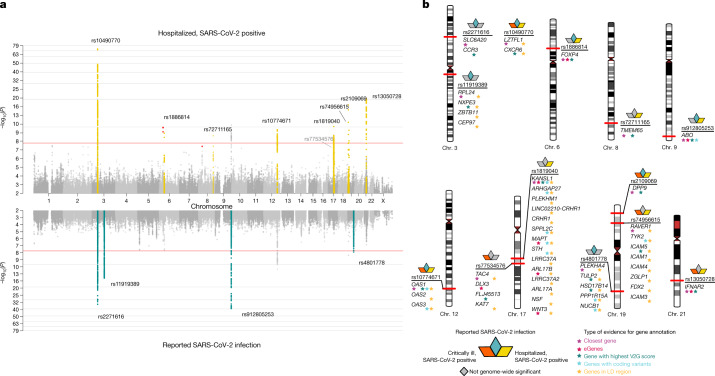
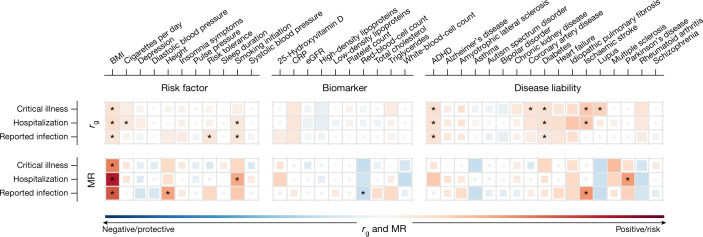
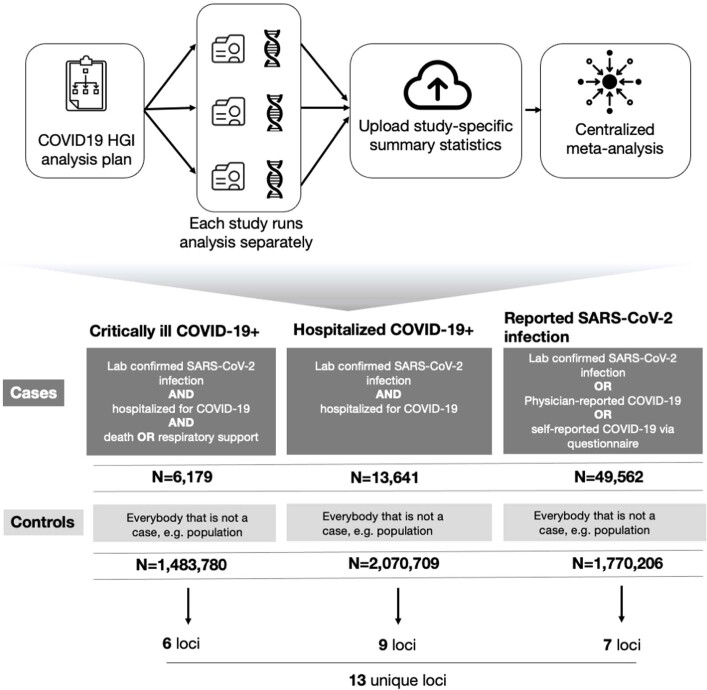
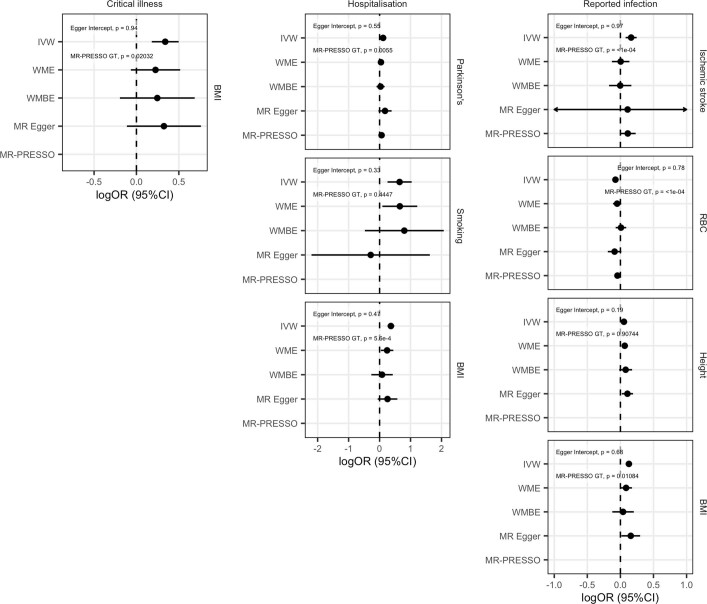
The genetic make-up of an individual contributes to the susceptibility and response to viral infection. Although environmental, clinical and social factors have a role in the chance of exposure to SARS-CoV-2 and the severity of COVID-191,2, host genetics may also be important. Identifying host-specific genetic factors may reveal biological mechanisms of therapeutic relevance and clarify causal relationships of modifiable environmental risk factors for SARS-CoV-2 infection and outcomes. We formed a global network of researchers to investigate the role of human genetics in SARS-CoV-2 infection and COVID-19 severity. Here we describe the results of three genome-wide association meta-analyses that consist of up to 49,562 patients with COVID-19 from 46 studies across 19 countries. We report 13 genome-wide significant loci that are associated with SARS-CoV-2 infection or severe manifestations of COVID-19. Several of these loci correspond to previously documented associations to lung or autoimmune and inflammatory diseases3-7. They also represent potentially actionable mechanisms in response to infection. Mendelian randomization analyses support a causal role for smoking and body-mass index for severe COVID-19 although not for type II diabetes. The identification of novel host genetic factors associated with COVID-19 was made possible by the community of human genetics researchers coming together to prioritize the sharing of data, results, resources and analytical frameworks. This working model of international collaboration underscores what is possible for future genetic discoveries in emerging pandemics, or indeed for any complex human disease.
© 2021. The Author(s).
Conflict of interest statement
A full list of competing interests is supplied as Supplementary Table 13.
Figures

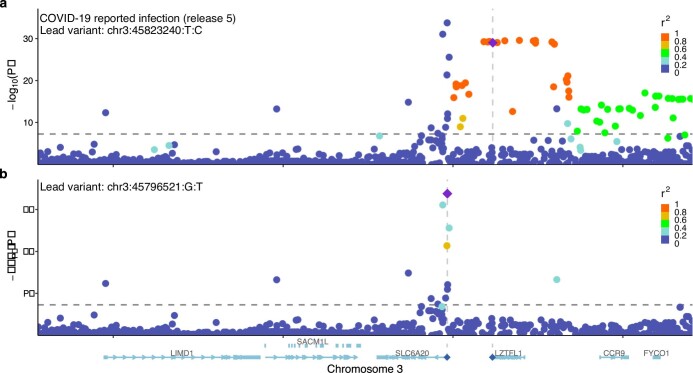
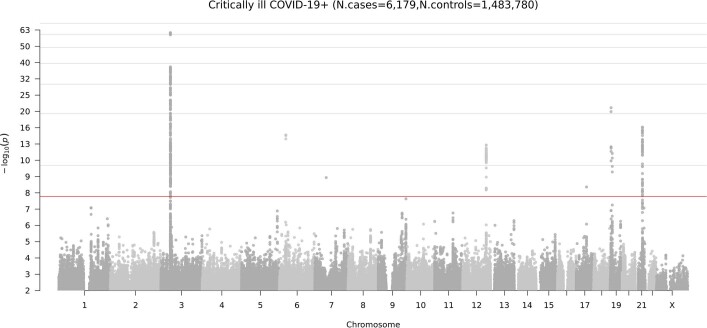
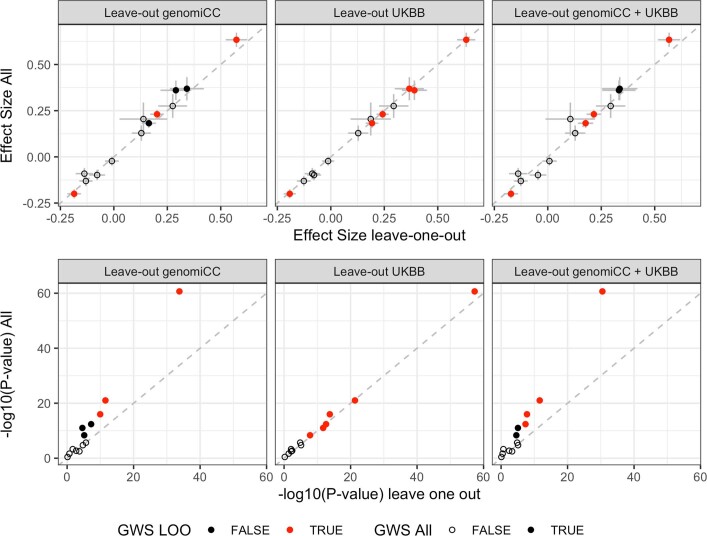
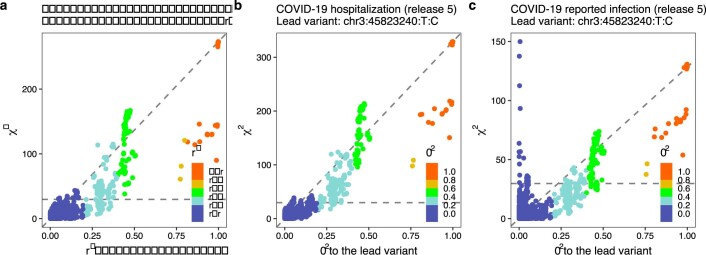
Comment in
-
Human genetic variants identified that affect COVID susceptibility and severity.Nature. 2021 Dec;600(7889):390-391. doi: 10.1038/d41586-021-01773-7. Nature. 2021. PMID: 34239107 No abstract available.
-
The quest to find genes that drive severe COVID.Nature. 2021 Jul;595(7867):346-348. doi: 10.1038/d41586-021-01827-w. Nature. 2021. PMID: 34239118 No abstract available.
-
A first update on mapping the human genetic architecture of COVID-19.Nature. 2022 Aug;608(7921):E1-E10. doi: 10.1038/s41586-022-04826-7. Epub 2022 Aug 3. Nature. 2022. PMID: 35922517 Free PMC article. No abstract available.
-
A second update on mapping the human genetic architecture of COVID-19.Nature. 2023 Sep;621(7977):E7-E26. doi: 10.1038/s41586-023-06355-3. Epub 2023 Sep 6. Nature. 2023. PMID: 37674002 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Grants and funding
- MR/L501542/1/MRC_/Medical Research Council/United Kingdom
- MR/R026408/1/MRC_/Medical Research Council/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_19025/MRC_/Medical Research Council/United Kingdom
- F31 MH124306/MH/NIMH NIH HHS/United States
- MR/S020039/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12014/12/MRC_/Medical Research Council/United Kingdom
- MC_UU_12014/10/MRC_/Medical Research Council/United Kingdom
- MR/P011705/2/MRC_/Medical Research Council/United Kingdom
- MR/S006753/1/MRC_/Medical Research Council/United Kingdom
- MC_UP_A090_1006/MRC_/Medical Research Council/United Kingdom
- G0901254/MRC_/Medical Research Council/United Kingdom
- MR/P011705/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/10/MRC_/Medical Research Council/United Kingdom
- MC_UU_12014/8/MRC_/Medical Research Council/United Kingdom
- K12 AR084232/AR/NIAMS NIH HHS/United States
- MC_UU_00007/15/MRC_/Medical Research Council/United Kingdom
- 210561/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- MR/K01417X/1/MRC_/Medical Research Council/United Kingdom
- G-0907/PUK_/Parkinson's UK/United Kingdom
- MR/S032304/1/MRC_/Medical Research Council/United Kingdom
- R01 GM133169/GM/NIGMS NIH HHS/United States
- G0701075/MRC_/Medical Research Council/United Kingdom
- MC_UU_12008/1/MRC_/Medical Research Council/United Kingdom
- R01 HL142302/HL/NHLBI NIH HHS/United States
- G-1307/PUK_/Parkinson's UK/United Kingdom
- MC_PC_20004/MRC_/Medical Research Council/United Kingdom
- MC_UU_00011/1/MRC_/Medical Research Council/United Kingdom
- T32 HG000040/HG/NHGRI NIH HHS/United States
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- MR/N02995X/1/MRC_/Medical Research Council/United Kingdom
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- K01 MH121659/MH/NIMH NIH HHS/United States
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_19026/MRC_/Medical Research Council/United Kingdom
- R01 HG009120/HG/NHGRI NIH HHS/United States
- MC_PC_19059/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
- Archivio Istituzionale della Ricerca Unimi - Access Free Full Text
- Europe PubMed Central
- Kyoto University Research Information Repository - Articles
- Nature Publishing Group
- ORBi (University of Liege)
- PubMed Central
- University of Turin Instituional Repository AperTO - Free full text
- White Rose Research Online
- eScholarship, University of California - Access Free Full Text
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
